
An efficient green protocol for the preparation of acetoacetamides and application of the methodology to a one-pot synthesis of Biginelli dihydropyrimidines. Expansion of dihydropyrimidine topological chemical space†
Abstract
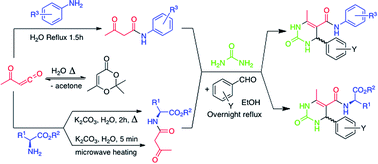
The present study describes the preparation of N-aryl-(15) and N-alkyl-(17) acetoacetamides, in good to excellent yields, using both conventional and microwave heating, by reaction of amine derivatives (14 and 16) with 2,2,6-trimethyl-4H-1,3-dioxin-4-one (TMD, 12) in aqueous medium. The acetoacetamides were used to prepare novel Biginelli dihydropyrimidine derivatives. The introduction of the amino acid derivatives potentially allows for the exploration of new structural complexity and topologically diversifies the chemical space occupied by this versatile chemical scaffold.
 Please wait while we load your content...
Please wait while we load your content...

