
In situ preparation of cobalt doped ZnO@C/CNT composites by the pyrolysis of a cobalt doped MOF for high performance lithium ion batteries†
Hongyun Yue*abc,
Zhenpu Shia,
Qiuxian Wanga,
Ting dua,
Yanmin Dinga,
Jun Zhanga,
Ningning Huoa and
Shuting Yang*abc
aSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, China
bEngineering Technology Research Center of Motive Power and Key Materials, Xinxiang, Henan 453007, China
cCollaborative Innovation Center of Henan Province for Motive Power and Key Materials, Xinxiang, Henan 453007, China. E-mail: yuehongyun@foxmail.com; shutingyang@foxmail.com; Fax: +86-373-3326439; Tel: +86-373-3326439
First published on 28th August 2015
Abstract
Co doped ZnO embedded in carbon/carbon nanotube composites (CZO@C/CNT) was prepared in situ during the calcination of Co-MOF-105 at 600 °C. A lower crystallinity demonstrated weaker binding force in Co-MOF-105, which made it possible for Co ions to break away from the crystal and reduce to metal Co during the pyrolysis process. The formation of CNTs was catalyzed by Co metal and the carbon source was terephthalic acid, which acted as the organic linker in the MOF. Moreover, the sp2 hybridization of the carbon atoms in terephthalic acid decreased the energy barrier during the growth of CNTs. From TEM and SEM observation, the CNTs were interspersed in the material and connected the CZO@C nanoparticles together, which made the electron transfer easier. The other advantages of Co doping were enhancing the conductivity of ZnO and increasing the graphitization degree of the carbon on the surface of the CZO@C nanoparticles. When the CZO@C/CNT composite was used as an anode material for lithium ion batteries, an enhanced electrochemical performance of 758 mA h g−1 after 100 cycles at a current density of 100 mA g−1 was obtained.
1. Introduction
Environmental pollution and the exhaustion of fossil fuel are two major problems facing human beings. An effective solution is to replace traditional motor vehicles with electric vehicles (EVs). However, the development of EVs is limited by the efficiency, security and cycle life of batteries.1–4 Lithium ion batteries (LIBs) are a promising candidate due to their high energy density and long cycle life. So, developing novel anode materials to meet the critical demand of EVs is a popular topic.5–7 As an anode material, ZnO has a higher theoretical lithium storage capacity (987 mA h g−1) than traditional graphite (372 mA h g−1) and a higher lithium ion diffusion coefficient compared to other transition metal oxides.8,9 In addition, ZnO is cheap and nontoxic.10 However, similar to other metal oxides, ZnO experiences volume expansion and poor electrical conductivity during cycling which results in the pulverization of electrodes and limited rate performance.11,12 The present solutions to overcome the disadvantages above consist of carbon coating,13 nanoparticle preparation,14 metal doping15 and oxide compositing.16 Doping and compositing conductive carbon materials,12,17 such as combining carbon nanotubes (CNTs)18 with metal oxides, have also attracted many researchers’ attention in recent years.Metal–organic frameworks (MOFs) are a class of rapidly growing adsorbent materials that have high surface area with high porosity.19,20 MOFs have been studied in many fields owing to their excellent properties, such as gas storage and separation,21 sensing or recognition,22 catalysis,23 electrochemistry,24 and so on. Presently, the application of MOFs in electrochemistry is still in its infancy, and they are most commonly used as precursors during the synthesis of nano-metal oxides or nanoporous carbon.25 The metal centers and functional linkers of MOFs are arranged with long range order. Ultra-small particles of metal oxides which are coated with carbon can be prepared by the simple pyrolysis of MOFs under an inert atmosphere.26 Metal ions can be easily absorbed by MOFs, which provides a simple way to prepare doped metal oxides.27 Porous carbon-coated ZnO quantum dots (QDs) derived from MOF exhibit nearly 100% capacity retention after 50 cycles.28 Zhang et al. developed a route to directly grow ZnO@ZnO QDs/C core–shell nanorod arrays (NRAs) on a flexible carbon cloth substrate, which can deliver a capacity retention of 699 mA h g−1 after 100 cycles at 500 mA g−1.29 Our previous work reported cobalt doped ZnO@C (CZO@C) which exhibited a capacity of 525 mA h g−1 up to the 50th cycle at a current density of 100 mA g−1.30
In this work, cobalt (Co) doped ZnO@C/CNT (CZO@C/CNT) composites were synthesized by the pyrolysis of a cobalt doped MOF (Co-MOF). At the best doping amount of 2%,30 the conductivity of ZnO was enhanced and the graphitization degree of the carbon on the surface of the CZO@C nanoparticles was increased, as proven by our previous work. Although enhanced performance was obtained in our previous work, the synthesis conditions were hard to control for the pyrolysis of the MOF by immediate cooling without holding at the maximum temperature. In the present work, the calcination conditions were easy to control. Additionally, the binding force between Co and the organic linker was weaker when the reaction temperature was 105 °C compared to that of 120 °C. Therefore, a small amount of Co ions was reduced to metal Co during the pyrolysis and acted as catalyst for the growth of CNTs. The CNTs were interspersed in the parent carbon which was embedded with ZnO nanoparticles and it was likely that the CNTs were conductive wires which linked the ZnO@C composites together. All of the three advantages of Co doping made electron transfer easier in the material. When the CZO@C/CNT composite was used as an anode material for lithium ion batteries, an enhanced electrochemical performance of 758 mA h g−1 after 100 cycles at a current density of 100 mA g−1 was obtained.
2. Experimental section
Material synthesis
Zinc nitrate hexahydrate (6.0 mmol) and terephthalic acid (2.0 mmol) were dissolved in 60 mL of N,N-dimethylformamide (DMF) in an autoclave. The reaction mixture was heated and maintained at 105 °C in an oven to yield cubic crystals of MOF-5. Cobalt nitrate hexahydrate (0.6 mmol) was added into the reaction system after the autoclave was cooled to room temperature. The mixture was stirred for 24 h in sealed surroundings and the reaction vessel was then kept at 105 °C for 24 h to yield Co-MOF-105 crystals. The obtained samples were washed repeatedly with ethanol and then dried in a vacuum oven at 60 °C. Co-MOF-120 was prepared as a contrast at a reaction temperature of 120 °C.For the synthesis of the CZO@C/CNT composite, the Co-MOF-105 was heat-treated at 600 °C for 2 h in a nitrogen atmosphere with a heating rate of 5 °C min−1. As a contrast, ZnO@C and CZO@C composites were synthesized by the pyrolysis of MOF-5 and Co-MOF-120 at 600 °C for 2 h in a nitrogen atmosphere.
Material characterization
Transmission electron microscopy (TEM, JOEL, JEM-2100), and field emission scanning electron microscopy (FESEM, Zeiss, JSM-6700F) were used to identify the morphological structures of the obtained samples. X-ray diffraction (XRD) was carried out on a D8 Advance (Bruker) diffractometer using Ni-filtered Cu Kα radiation (λ = 0.154184 nm). The powder samples were scanned from 10° to 80° under the operation conditions of 40 kV and 40 mA. X-ray photoelectron spectroscopy (XPS, PHI 5700 ESCA) analysis was performed using monochromated Al Kα radiation (hν = 1486.6 eV) to detect the existing state of the elements. Raman scattering measurements (Jobin Yvon HR800, HORIBA) were performed in a backscattering geometry at room temperature with a 514.5 nm line of an Ar-ion laser to measure the graphitization degree of the carbon in the as-synthesized composites. The specific surface area and pore size distribution were measured using the Brunauer–Emmet–Teller method (BET, Tristar-II 3020, Micromeritics).Electrochemical measurements
For the electrochemical evaluation of ZnO@C, CZO@C and CZO@C/CNT, the test electrodes consisted of the active powder material (60 wt%), conductive carbon black (20 wt%) as a conductor, and poly(vinylidene fluoride) (PVDF, 20 wt%) dissolved in N-methyl pyrrolidinone (NMP) as a binder. Each component was mixed well to form a slurry coating on a copper foil substrate. The active material loading on each disk was ∼1.7 mg, corresponding to ∼1.5 mg cm−2. Laboratory made, CR2032 type coin cells were assembled in an Ar-filled glove-box using Celgard 2400 as the separator, Li foil as the counter and reference electrodes, and 1 M LiPF6 in ethylene carbonate (EC)–dimethyl carbonate (DMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume) as the electrolyte. The coin cells were tested between 0.01 and 3.0 V (vs. Li/Li+) at a current density of 100 mA g−1 using a program controlled battery test instrument (LAND CT2001A, China). The gravimetric capacity was calculated with respect to all active elements (ZnO and C). Electrochemical impedance spectra (EIS) were conducted with a Solartron SI1287+SI1260 potentiometer at 25 °C with the frequency ranging from 100 kHz to 0.1 Hz.
:1 volume) as the electrolyte. The coin cells were tested between 0.01 and 3.0 V (vs. Li/Li+) at a current density of 100 mA g−1 using a program controlled battery test instrument (LAND CT2001A, China). The gravimetric capacity was calculated with respect to all active elements (ZnO and C). Electrochemical impedance spectra (EIS) were conducted with a Solartron SI1287+SI1260 potentiometer at 25 °C with the frequency ranging from 100 kHz to 0.1 Hz.
3. Results and discussion
The TEM image of the CZO@C/CNT composites is shown in Fig. 1a. A large number of CNTs were found in the CZO@C/CNT composites and there were no CNTs in the ZnO@C and CZO@C composites (Fig. S1a and b in the ESI†). Moreover, the CNTs were interspersed within the composites (Fig. 1b). The CNTs were multi-walled carbon nanotubes (MWCNTs) with a thickness of about 10 nm (Fig. 1c) and a diameter ranging from 20 to 60 nm. Additionally, the CNTs were connected with the carbon layer of CZO. The HRTEM image of CZO@C/CNTs is shown in Fig. 1d. The interplanar spacing measured from the image of 0.284 nm belongs to the (100) plane of ZnO (JCPDS card no. 89-7102) and the value of 0.336 nm belongs to the (002) plane of carbon (JCPDS card no. 75-0444). It was clear that the ZnO was covered by amorphous carbon and connected with the CNTs. The endpoint of the CNTs was also observed using HRTEM (Fig. 1e and f). In this work, a small amount of Co ions was reduced to metal Co during the pyrolysis and acted as catalyst for the growth of CNTs. The nanoparticles at the endpoint of the CNTs consisted of Co element (Fig. S2†) and the interplanar spacing was about 0.206 nm which belongs to the (002) plane of Co (JCPDS card no. 89-7373) in Fig. 1f. The carbon source was terephthalic acid which acted as an organic linker in the MOF. Moreover, the sp2 hybridization of the carbon atoms in terephthalic acid decreased the energy barrier during the CNT growth. | ||
| Fig. 1 TEM (a and b) and HRTEM (c–f) images of CZO@C/CNTs. | ||
The SEM images of the as-synthesized MOFs and ZnO based materials are shown in Fig. 2. All of the MOFs including MOF-5, Co-MOF-120 (Fig. 2a) and Co-MOF-105 (Fig. 2b) exhibited well defined cube morphology. Additionally, the surface of the MOF which grew at 120 °C (Fig. S3a†) was smoother than that at 105 °C (Fig. S3b†). After calcination, the cube morphology which was composed of nanoparticles was retained (Fig. S4a and b†). The primary nanoparticles of CZO@C and CZO@C/CNT had a similar diameter of about 20 nm (Fig. 2c and d). In addition, the smoother surface of Co-MOF-120 resulted in the nanoparticles packing closer in CZO@C. CZO@C/CNT presented porous structures with CNTs interspersed in the secondary particles. The porous structures made the contact of CZO@C and the electrolyte more convenient, and the conductive CNTs connected the CZO@C nanoparticles together which made electron transfer easier. The two advantages mentioned above could enhance the electrochemical performance to a great extent.
 | ||
| Fig. 2 SEM images of Co-MOF-120 (a), Co-MOF-105 (b), CZO@C (c) and CZO@C/CNT (d). | ||
The XRD patterns of the as-synthesized Co-MOFs are shown in Fig. 3a. Both Co-MOF-105 and Co-MOF-120 exhibited similar peak positions with the previously reported values of MOF-5 moieties. No new peaks appeared and no peaks shifted in the pattern of the Co-MOFs compared with the reported MOF-5.31 Obviously, the binary metal based MOF was formed (Fig. 3a). The difference between Co-MOF-105 and Co-MOF-120 was the degree of crystallization. The lower reaction temperature resulted in a lower degree of crystallization. Combined with the SEM images, it was confirmed that more crystal defects were formed in Co-MOF-105. The lower crystallinity demonstrated a weaker binding force in Co-MOF-105, which made it possible for Co ions to break away from the crystal and reduce to metal Co during the pyrolysis process. The major calcined product of MOF-5 in the N2 atmosphere was carbon coated wurtzite ZnO (JCPDS card no. 89-7102) (Fig. 3b). The peaks of CZO@C and CZO@C/CNT were a little shifted to the low angle region and no new peak appeared which indicated that the Co was doped in ZnO interstitially. The inset is the enlarged view of the ZnO based materials between 20° and 30°. The inconspicuous peak at 26° in CZO@C/CNT belongs to the diffraction peak of CNTs (Fig. 3b inset).
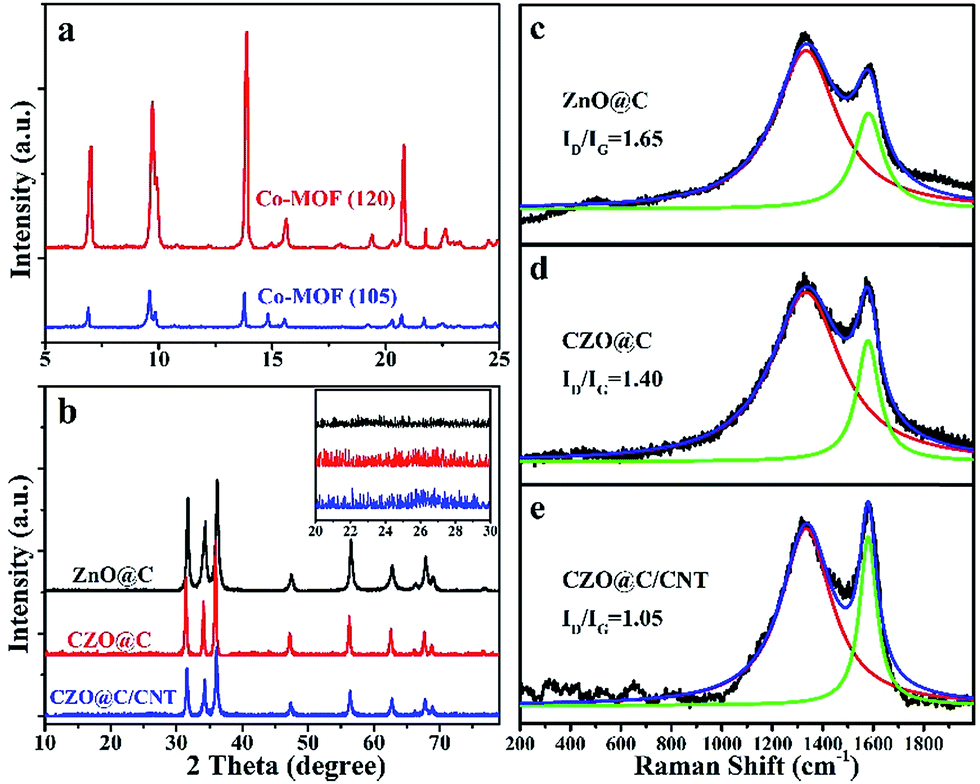 | ||
| Fig. 3 XRD patterns of the as synthesized MOFs (a) and ZnO based materials (b); Raman shift of as synthesized ZnO@C (c), CZO@C (d) and CZO@C/CNT (e). | ||
The Raman shift of the as-synthesized ZnO@C, CZO@C and CZO@C/CNT are shown in Fig. 3c–e, respectively. In these figures, two broad peaks at 1333.3 and 1581.1 cm−1 were clearly observed, which could be marked as the D and G bands of carbon, respectively.32 The ID/IG ratios of ZnO@C, CZO@C and CZO@C/CNT were 1.65, 1.40 and 1.05, respectively. The doped Co enhanced the graphitization degree of the carbon during the calcining process,30 which resulted in a lower ID/IG ratio of CZO@C compared to ZnO@C. The lowest ID/IG ratio was 1.05, which was attributed to the in situ formed CNTs in CZO@C/CNT.
In the C 1s core level XPS spectrum (Fig. 4a and b), the peaks at 284.5 eV, 285.3 eV, 286.7 eV and 289.0 eV belong to the bonds of C–C,33 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C,34 C–O35 and O–CO,35 respectively. The increased relative intensity of the peak at 285.3 eV confirms that most carbon atoms were sp2 hybridized in CZO@C/CNT, which was in accordance with the Raman shift. Two peaks at 781.1 eV for Co 2p3/2 and 796.8 eV for Co 2p1/2 were observed in Fig. 4c and d. The energy difference was 15.4 eV, which showed that the major existence state of Co was the Co(II) oxidation state.36 Moreover, the low intensity of the curve was caused by the low content of Co in the composites. The peaks at 786.8 and 802.5 eV were ascribed to the satellite peaks. The binding energies of the Zn 2p3/2 peak and Zn 2p1/2 peak of ZnO@C were at 1020.7 eV and 1043.8 eV, indicating that the existence state of the Zn element was the Zn(II) oxidation state37 (Fig. S5a†). However, the relative peaks of CZO@C and CZO@C/CNT were a little positively shifted in binding energy (Fig. S5b and c†), which was caused by the Co doping. The result was in accordance with the XRD patterns.
C,34 C–O35 and O–CO,35 respectively. The increased relative intensity of the peak at 285.3 eV confirms that most carbon atoms were sp2 hybridized in CZO@C/CNT, which was in accordance with the Raman shift. Two peaks at 781.1 eV for Co 2p3/2 and 796.8 eV for Co 2p1/2 were observed in Fig. 4c and d. The energy difference was 15.4 eV, which showed that the major existence state of Co was the Co(II) oxidation state.36 Moreover, the low intensity of the curve was caused by the low content of Co in the composites. The peaks at 786.8 and 802.5 eV were ascribed to the satellite peaks. The binding energies of the Zn 2p3/2 peak and Zn 2p1/2 peak of ZnO@C were at 1020.7 eV and 1043.8 eV, indicating that the existence state of the Zn element was the Zn(II) oxidation state37 (Fig. S5a†). However, the relative peaks of CZO@C and CZO@C/CNT were a little positively shifted in binding energy (Fig. S5b and c†), which was caused by the Co doping. The result was in accordance with the XRD patterns.
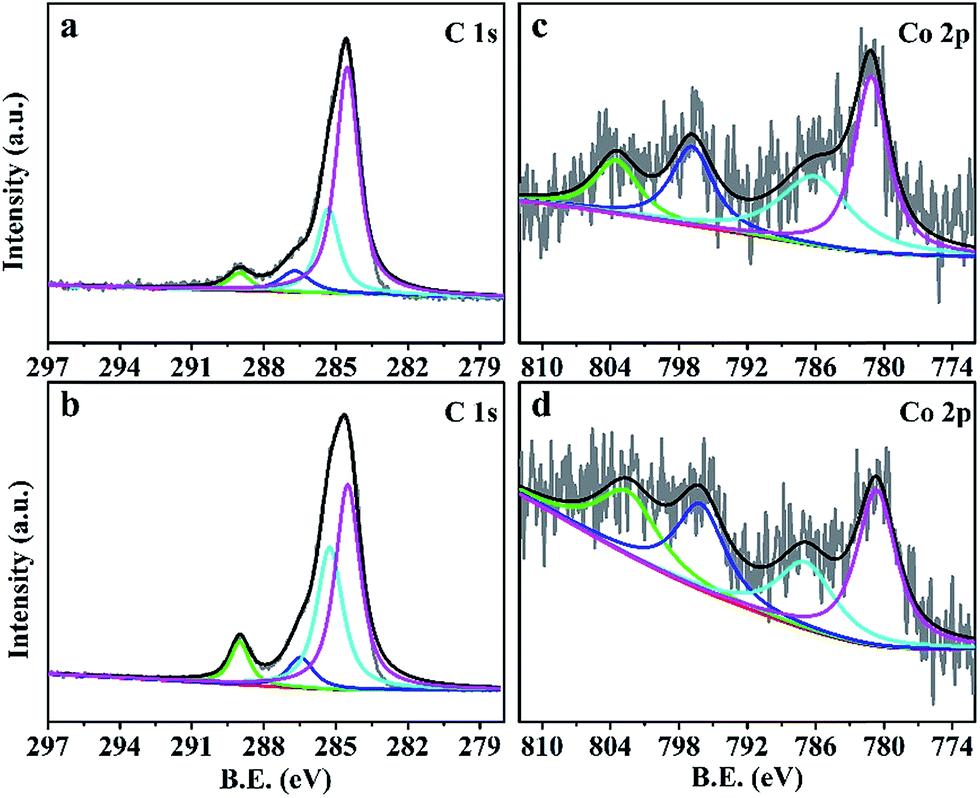 | ||
| Fig. 4 C 1s core level XPS profile spectra of CZO@C (a) and CZO@C/CNT (b); Co 2p core level XPS profile spectra of CZO@C (c) and CZO@C/CNT (d). | ||
N2 adsorption–desorption measurements were taken to characterize the specific surface area and pore size distribution of CZO@C/CNT. As shown in Fig. S6a,† a type IV isotherm with an inconspicuous hysteresis loop in the middle pressure region occurs, indicating mesoporous characteristics. The Brunauer–Emmett–Teller (BET) surface areas and total pore volume of CZO@C/CNT were 102.8 m2 g−1 and 0.31 cm3 g−1, respectively. The pore size distribution calculated using the Barrett–Joyner–Halenda (BJH) method showed that the CZO@C/CNT were mesoporous composites with a wide distribution centered at 40 nm, and also some macropores were present (Fig. S6b†).
The initial discharge capacities of the CZO@C/CNT, CZO@C and ZnO@C composites were 1551, 880 and 903 mA h g−1, respectively (Fig. 5a–c). These discharge capacities became 1091, 640 and 665 mA h g−1 for the second cycle. The charge and discharge capacity of CZO@C/CNT for the 10th cycle was almost the same as the values for the 50th cycle, which showed the best stability of the CZO@C/CNT composites. These high irreversible capacities at the first cycle were caused by the formation of a solid electrolyte interphase (SEI) layer on the surface of the electrode.29,38 The capacity decay was quite large for the first five cycles. One reason was the morphological change during the formation of the LixZn alloys and the other was the poor conductivity of ZnO.39 The CZO@C/CNT composites exhibited enhanced electrochemical performance compared with ZnO@C and CZO@C composites (Fig. 5d). The CZO@C/CNT composites had a high discharge capacity of 758 mA h g−1 after 100 cycles, which was much higher than that of CZO@C (523 mA h g−1 after 100 cycles) and ZnO@C (335 mA h g−1 after 50 cycles). The improved performance of CZO@C/CNT compared with CZO@C was attributed to the CNTs in the composites. A conductive network was formed and the conductivity of the material was enhanced, which was attributed to the easier electron transfer in the material. The reactions of CZO with Li were similar to that of ZnO, including the reversible conversion of metal oxide to nanosized metal and Li oxide matrix, and the alloying and dealloying processes of metal and Li.40,41 The reactions were as follows:
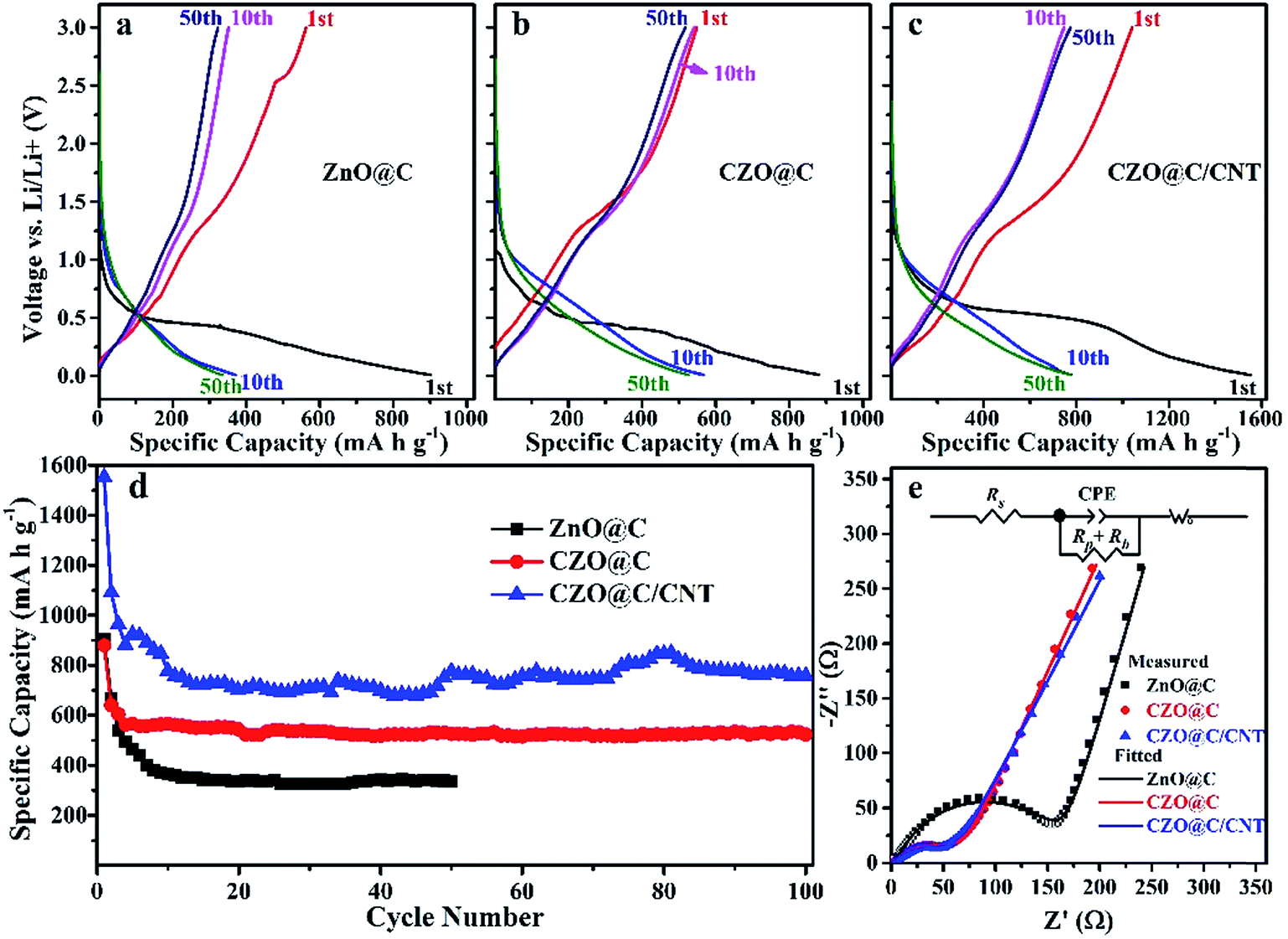 | ||
| Fig. 5 Charge/discharge curve of ZnO@C (a), CZO@C (b), CZO@C/CNT (c), cycling performance (d) and Nyquist plots of the three ZnO based materials after 50 cycles (e). | ||
Discharge process:
| CZO + 2Li+ + 2e− → Zn + Co + Li2O | (1) |
| Zn + Li+ + e− → LiZn | (2) |
| Co + Li+ + e− → LiCo | (3) |
Charge process:
| LiZn → Zn + Li+ + e− | (4) |
| LiCo → Co + Li+ + e− | (5) |
| Zn + Li2O → ZnO + 2Li+ + 2e− | (6) |
| Co + Li2O → CoO + 2Li+ + 2e− | (7) |
The EIS experiments were carried out (Fig. 5e) to further understand the improved Li storage performance of the CZO@C/CNT composites. All of the Nyquist plots showed depressed separate semicircles in the middle and high frequency region and a straight line in the low frequency region. The semicircles corresponded to the charge-transfer resistance to Li ions at the interface between the electrode and electrolyte (Rp) and the electronic resistivity of the active material and ionic conductivity in the electrode (Rb).12,13 The straight line was assigned to the Warburg impedance (Wo) corresponding to the Li diffusion process,42 and Rs represented the internal resistance of the test battery. As shown in Fig. 5e, the CZO based composites showed a distinctly smaller semicircle, indicating that they had lower charge-transfer impedance and electronic resistivity than that of the ZnO@C composites owing to that Co doping could enhance the conductivity of ZnO. The tiny conduction improvement of CZO@C/CNT was attributed to the CNTs in the composite.
Finally, the capacities presented in this work were compared with those taken from the literature for various electrodes made from ZnO based anode materials, as shown in Table 1.
| Electrode material | Reversible capacity (mA h g−1) | Cycle number | Voltage (V) | Rate (mA g−1) | Ref. |
|---|---|---|---|---|---|
| a Graphene-encapsulated porous carbon-ZnO. | |||||
| Ultrathin ZnO nanotubes | 386 | 50 | 0.01–2.5 | 494 | 43 |
| ZnO/ZnCo2O4 rods | 900 | 30 | 0.01–3.0 | 45 | 41 |
| Ni-coated ZnO | 490 | 30 | 0.02–3.0 | 80 | 39 |
| ZnO–Ag–C | 729 | 200 | 0.01–3.0 | 100 | 15 |
| ZnO@C-5 | 518 | 300 | 0.02–3.0 | 110 | 13 |
| G-C8-ZnOa | 560 | 100 | 0.01–3 | 97.8 | 17 |
| ZnO@ZnO QDs/C NRAs | 699 | 100 | 0.01–2 | 500 | 29 |
| ZnO-M/PC | 653 | 100 | 0.1–3.0 | 100 | 38 |
| CZO@C | 523 | 100 | 0.01–3.0 | 100 | This work |
| CZO@C/CNT | 758 | 100 | 0.01–3.0 | 100 | This work |
4. Conclusions
CNTs were grown out in situ in CZO@C/CNT during the calcination of Co-MOF-105 at 600 °C. The catalyst was metal Co and the carbon source was terephthalic acid, which acted as an organic linker in the MOF. Moreover, the sp2 hybridization of the carbon atoms in terephthalic acid decreased the energy barrier during the CNT growth. TEM and SEM observation found that the CNTs were interspersed in the material and connected the CZO@C nanoparticles together. The other advantages of Co doping were the enhancement of the conductivity of ZnO and increasing the graphitization degree of the carbon on the surface of the CZO@C nanoparticles. All of the three advantages of Co doping made electron transfer easier in the material. When the CZO@C/CNT composite was used as an anode material for LIBs, enhanced electrochemical performance of 758 mA h g−1 after 100 cycles at a current density of 100 mA g−1 was obtained.Acknowledgements
This work was financially supported by the Research Fund for the Doctoral Program of Higher Education of China (No. qd12124), Major Science and Technology Projects in Henan Province (No. 121100210500) and Key Project of Science and Technology of the Henan Educational Committee (No. 13A150518).References
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- K. Amine, R. Kanno and Y. H. Tzeng, MRS Bull., 2014, 39, 395–405 CrossRef CAS.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- M. V. Reddy, G. V. S. Rao and B. V. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- A. D. Roberts, X. Li and H. Zhang, Chem. Soc. Rev., 2014, 43, 4341–4356 RSC.
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454–2484 CAS.
- G. Z. Yang, H. W. Song, H. Cui, Y. C. Liu and C. X. Wang, Nano Energy, 2013, 2, 579–585 CrossRef CAS PubMed.
- J. Xie, N. Imanishi, A. Hirano, Y. Takeda, O. Yamamoto, X. B. Zhao and G. S. Cao, Thin Solid Films, 2011, 519, 3373–3377 CrossRef CAS PubMed.
- V. Aruoja, H. C. Dubourguier, K. Kasemets and A. Kahru, Sci. Total Environ., 2009, 407, 1461–1468 CrossRef CAS PubMed.
- X. Sun, C. G. Zhou, M. Xie, H. T. Sun, T. Hu, F. Y. Lu, S. M. Scott, S. M. George and J. Lian, J. Mater. Chem. A, 2014, 2, 7319–7326 CAS.
- C. T. Hsieh, C. Y. Lin, Y. F. Chen and J. S. Lin, Electrochim. Acta, 2013, 111, 359–365 CrossRef CAS PubMed.
- Z. M. Ren, Z. Y. Wang, C. Chen, J. Wang, X. X. Fu, C. Y. Fan and G. D. Qian, Electrochim. Acta, 2014, 146, 52–59 CrossRef CAS PubMed.
- Y. Hwa, J. H. Sung, B. Wang, C. M. Park and H. J. Sohn, J. Mater. Chem., 2012, 22, 12767–12773 RSC.
- Q. Xie, Y. Ma, D. Zeng, X. Zhang, L. Wang, G. Yue and D. L. Peng, ACS Appl. Mater. Interfaces, 2014, 6, 19895–19904 CAS.
- L. J. Liu, C. Y. Zhao, H. L. Zhao, Q. Y. Zhang and Y. Li, Electrochim. Acta, 2014, 135, 224–231 CrossRef CAS PubMed.
- R. Guo, W. B. Yue, Y. M. An, Y. Ren and X. Yan, Electrochim. Acta, 2014, 135, 161–167 CrossRef CAS PubMed.
- S. M. Abbas, S. T. Hussain, S. Ali, N. Ahmad, N. Ali and S. Abbas, J. Mater. Sci., 2013, 48, 5429–5436 CrossRef CAS.
- S. L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 CAS.
- W. Xuan, C. Zhu, Y. Liu and Y. Cui, Chem. Soc. Rev., 2012, 41, 1677–1695 RSC.
- T. A. Makal, J. R. Li, W. Lu and H. C. Zhou, Chem. Soc. Rev., 2012, 41, 7761–7779 RSC.
- H. L. Jiang, Y. Tatsu, Z. H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586–5587 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- L. Yang, S. Kinoshita, T. Yamada, S. Kanda, H. Kitagawa, M. Tokunaga, T. Ishimoto, T. Ogura, R. Nagumo, A. Miyamoto and M. Koyama, Angew. Chem., Int. Ed., 2010, 49, 5348–5351 CrossRef CAS PubMed.
- W. Yin, Y. Shen, F. Zou, X. Hu, B. Chi and Y. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 4947–4954 CAS.
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26, 6622–6628 CrossRef CAS PubMed.
- D. K. Lee, J. H. Park, J. I. Choi, Y. Lee, S. J. Kim, G. H. Lee, Y. H. Kim and J. K. Kang, Nanoscale, 2014, 6, 10995–11001 RSC.
- S. J. Yang, S. Nam, T. Kim, J. H. Im, H. Jung, J. H. Kang, S. Wi, B. Park and C. R. Park, J. Am. Chem. Soc., 2013, 135, 7394–7397 CrossRef CAS PubMed.
- G. Zhang, S. Hou, H. Zhang, W. Zeng, F. Yan, C. C. Li and H. Duan, Adv. Mater., 2015, 27, 2400–2405 CrossRef CAS PubMed.
- H. Y. Yue, Z. P. Shi, Q. X. Wang, Z. X. Cao, H. Y. Dong, Y. Qiao, Y. H. Yin and S. T. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 17067–17074 CAS.
- J. Hafizovic, M. Bjorgen, U. Olsbye, P. D. C. Dietzel, S. Bordiga, C. Prestipino, C. Lamberti and K. P. Lillerud, J. Am. Chem. Soc., 2007, 129, 3612–3620 CrossRef CAS PubMed.
- Z. Q. Zhu, S. W. Wang, J. Du, Q. Jin, T. R. Zhang, F. Y. Cheng and J. Chen, Nano Lett., 2014, 14, 153–157 CrossRef CAS PubMed.
- Y. Liu, W. Wang, L. Gu, Y. W. Wang, Y. L. Ying, Y. Y. Mao, L. W. Sun and X. S. Peng, ACS Appl. Mater. Interfaces, 2013, 5, 9850–9855 CAS.
- A. J. Maxwell, P. A. Brühwiler, A. Nilsson, N. Mårtensson and P. Rudolf, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 10717–10725 CrossRef CAS.
- H. L. Poh, P. Simek, Z. Sofer and M. Pumera, ACS Nano, 2013, 7, 5262–5272 CrossRef CAS PubMed.
- S. Y. Yang, R. Q. Lv, C. Z. Wang, Y. Y. Liu and Z. Q. Song, J. Alloys Compd., 2013, 579, 628–632 CrossRef CAS PubMed.
- T. F. Hung, S. G. Mohamed, C. C. Shen, Y. Q. Tsai, W. S. Chang and R. S. Liu, Nanoscale, 2013, 5, 12115–12119 RSC.
- X. Shen, D. Mu, S. Chen, B. Wu and F. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 3118–3125 CAS.
- C. Q. Zhang, J. P. Tu, Y. F. Yuan, X. H. Huang, X. T. Chen and F. Mao, J. Electrochem. Soc., 2007, 154, A65–A69 CrossRef CAS PubMed.
- H. Q. Dai, H. Xu, Y. N. Zhou, F. Lu and Z. W. Fu, J. Phys. Chem. C, 2012, 116, 1519–1525 CAS.
- L. L. Hu, B. H. Qu, C. C. Li, Y. J. Chen, L. Mei, D. N. Lei, L. B. Chen, Q. H. Li and T. H. Wang, J. Mater. Chem. A, 2013, 1, 5596–5602 CAS.
- L. Chen, H. Y. Xu, L. Li, F. F. Wu, J. Yang and Y. T. Qian, J. Power Sources, 2014, 245, 429–435 CrossRef CAS PubMed.
- K. T. Park, F. Xia, S. W. Kim, S. B. Kim, T. Song, U. Paik and W. I. Park, J. Phys. Chem. C, 2013, 117, 1037–1043 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra14271g |
| This journal is © The Royal Society of Chemistry 2015 |