
Anti-inflammatory alkaloid glycoside and quinoline alkaloid derivates from the stems of Clausena lansium†
Jie Liu,
Chuang-Jun Li,
Lin Ni,
Jing-Zhi Yang,
Li Li,
Cai-xia Zang,
Xiu-Qi Bao,
Dan Zhang and
Dong-Ming Zhang*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, People's Republic of China. E-mail: zhangdm@imm.ac.cn; Fax: +86-10-63165227; Tel: +86-10-63165227
First published on 16th September 2015
Abstract
Six new alkaloid glycosides, Clausenasides A–F (1–6), along with two new quinoline alkaloids, Clausenasides G–H (7–8), and ten known compounds (9–18) were obtained from the stems of C. lansium. The structures of the new compounds were elucidated on the basis of their spectroscopic analysis, and the absolute configurations of 1, 2, 3 and 7 were confirmed by Mosher's method, CD and ECD spectra, respectively. Compounds 4, 6, 9, 17, and 18 showed moderate inhibitory effects on LPS-induced NO production in murine microglial BV2 cells (IC50 values < 10 μM).
Introduction
Clausena lansium (Lour.) skeels (Rutaceae), a fruit tree, was widely distributed in southern China. In traditional Chinese medicine, the leaves and roots of C. lansium were used for coughs, asthma, dermatological disease, viral hepatitis, and gastro-intestinal diseases; the seeds are used to treat acute and chronic gastro-intestinal inflammation, ulcers, etc.1 Various bioactive constituents including coumarins, carbazole alkaloids and amide alkaloids have been isolated and identified from this plant.2–4 Previously, twenty new natural products including thirteen new carbazole alkaloids,5,6 four new coumarins,7 a new amide and a new megastigmane glucoside8 from the leaves and skeels of C. lansium were reported by our research group, and some of these alkaloids showed selective neuroprotective effects. In order to continue research for microscale bioactive metabolites from C. lansium, 200 kg stems of C. lansium were extracted by EtOAc and n-BuOH. This paper reported further investigation of n-BuOH extract from the stems of C. lansium which led to the isolation and characterization of six new alkaloid glycosides (1–6), two new quinoline alkaloids (7–8) along with ten known compounds (9–18) from the stems of C. lansium (Fig. 1). It was the first time to obtain these alkaloid glycosides from C. lansium. The inhibitory effects on LPS-induced NO production in a murine microglial cell lines of 1–18 were also evaluated. We present herein the isolation and structural characterization of Clausenasides A–H, as well as their bioactivities. | ||
| Fig. 1 Alkaloid derivates (1–18) obtained from the stems of C. lansium. | ||
Results and discussion
Clausenaside A (1) was obtained as a white powder. Its molecular formula was assigned as C22H27NO8 based on the 13C NMR spectroscopic data and HRESIMS (m/z 456.1634 [M + Na]+, calcd for C22H27NO8Na, 456.1629), implying ten indices of hydrogen deficiency. The IR spectrum displayed absorptions characteristic of amino (3315 cm−1), amide (1640 cm−1) and aromatic ring (1618, 1579 and 1505 cm−1) groups, and the UV spectrum showed absorptions at λmax 202 and 226 nm. A 1,4-disubstituted benzene ring at δH 7.32 (2H, d, J = 8.8 Hz), 6.87 (2H, d, J = 8.8 Hz) and a 1-substituted benzene ring at δH 7.79 (2H, d, J = 7.2 Hz), 7.51 (1H, m), 7.44 (2H, t, J = 7.6 Hz) were clearly observed in the 1H NMR spectrum and were assigned with the aid of COSY correlations (Fig. 2). In addition, an amide group δH 8.33 (1H, t, J = 4.0 Hz, NH), a methoxy group [δH 3.73; δC 55.0], a methylene group [δH 3.69 (1H, m), 3.55 (1H, m); δC 44.5], an oxygenated methine group [δH 4.83 (1H, t, J = 4.0 Hz); δC 78.8], and a β-glucopyranosyl moiety [4.42 (1H, d, J = 7.6 Hz), δH 3.06–3.56 (5H, m); δC 102.9, 73.8, 76.5, 69.9, 76.8, 60.9] were clearly observed in the 1H and 13C NMR spectra along with HSQC correlations. On the basis of the NMR techniques, compound 1 was elucidated as an amide alkaloid glycoside. In the HMBC spectrum (Fig. 2), the cross-peaks between H-3, H-5/C-1 (δC 134.4), H-2, H-6/C-7 (δC 166.2) demonstrated the 1-substituted benzene ring attached to the carbonyl group. H-8/C-7 (δC 166.2), N-H/C-7 (δC 166.2), C-9 (δC 78.8) indicated the methylene group attached to the N atom. H-9/C-10 (δC 132.5), C-11 (δC 127.9), H-1′/C-9 (δC 78.8) implying that 1,4-disubstituted benzene ring and β-glucopyranosyl unit were linked to C-9. The methoxy group resonated at C-13 (δC 158.5). Hydrolysis of 1 with snailase produced the aglycone (1a) and glucose. 1a was identified as tembamide9 by comparison of its 1H and 13C NMR, and HRESIMS. The absolute configuration o of 1a was determined as 9S by comparison of the optical rotation of 1a {[α]25D +54.8 (c 0.20 CHCl3)} and (S)-(+)-tembamide10 {[α]25D +56.9 (c 0.54 CHCl3)}. Compound 1a was treated with (R)- and (S)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (MTPA-Cl) in anhydrous CH2Cl2 (ref. 11) to afforded the 1a-(S)-MTPA ester (1aa) and 1a-(R)-MTPA ester (1ab), respectively. The ΔδSRH values were calculated as shown in Fig. 3. Application of Mosher's rule12 revealed that 1a had the 9S configuration. The CD spectrum of 1a exhibited negative Cotton effect at 235 nm (Fig. 4), and the CD spectrum of 1 also showed negative Cotton effect at 238 nm (Fig. 4), which indicated that the structure of 1a had not changed in the process of enzymatic hydrolysis and the absolute configuration of 1 was elucidated as 9S. The D-glucose was identified by comparison of TLC and optical rotation data of D-glucose and the authentic sample.13 Thus, the structure of 1 was defined as (S)-(+)-tembamide-9-O-β-D-glucopyranoside.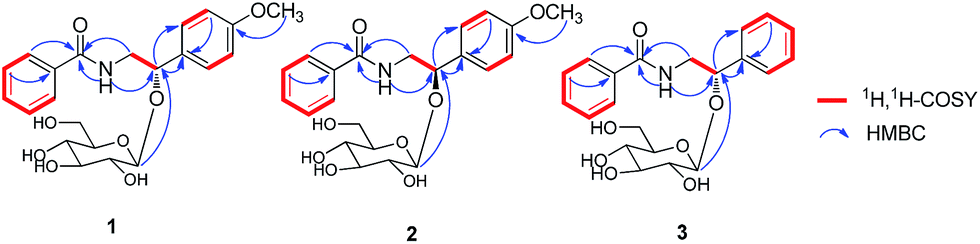 | ||
| Fig. 2 Key 1H, 1H-COSY and HMBC correlations of compounds 1, 2 and 3. | ||
 | ||
| Fig. 3 The Mosher's method of 1a. | ||
 | ||
| Fig. 4 The experimental CD spectrum of 1, 2, 3 and 1a. | ||
Clausenaside B (2) was obtained as a white powder. Its molecular formula was established as C22H27NO8 from the 13C NMR and positive-ion HRESIMS (m/z 456.1635 [M + Na]+, calcd for C22H27NO8Na, 456.1629), indicating ten indices of hydrogen deficiency. Comparison of spectroscopic data of 2 with those of 1 revealed that the planar structure of 2 was established the same as that of 1. Therefore, compound 2 was defined to be a stereo-isomer of 1. The CD spectra of 2 which showed positive Cotton effect at 237 nm (Fig. 4) was contrary to 1. Thus, the structure of 2 was established as (R)-(−)-tembamide-9-O-β-D-glucopyranoside.
Clausenaside C (3) was obtained as a white powder. Its molecular formula was deduced as C21H25NO7 on the basis of its 13C NMR and HRESIMS at m/z 426.1524 [M + Na]+, calcd for C21H25NO7Na, 426.1523, implying ten indices of hydrogen deficiency. Comparison of the spectroscopic data of 3 with those of 1 revealed that compound 3 also was an amide alkaloid glycoside, with the primary difference being the absence of a methoxy group at C-13 (δC 127.8). Compound 3 showed similar CD Cotton effects to those of 1 (Fig. 4), and the absolute configuration of 3 was 9S. Thus, the structure of 3 was defined as (S)-(+)-demethoxy-tembamide-9-O-β-D-glucopyranoside.
Clausenaside D (4) was obtained as a white powder. Its molecular formula was determined as C16H19NO7 based on its 13C NMR spectroscopic and HRESIMS at m/z 338.1232 [M + H]+ (calcd for C16H20NO7, 338.1234), corresponding with eight indices of hydrogen deficiency. The UV spectrum showed absorptions at 226, 270 and 322 nm. The 1H NMR spectrum (Table 2) showed o-substituted benzene ring [δH 8.04 (1H, dd, J = 7.8, 1.8 Hz, H-5); 7.28 (1H, dt, J = 7.8, 0.6 Hz, H-6); 7.66 (1H, dt, J = 8.4, 1.8 Hz, H-7); 7.52 (1H, d, J = 8.4 Hz, H-8)], a bond group δH 6.19 (1H, s, H-3), a methyl group δH 3.57, and a β-glucopyranosyl moiety δH 5.09 (1H, d, J = 7.8 Hz, H-1′). The 13C NMR spectrum (Table 2) exhibited 16 carbon signals including six in glucopyranosyl unit, a methyl group, and the remaining nine in the quinoline moiety. A comparison of the 1H and 13C NMR of 4 with those of integriquinolone14 suggested that their structures are closely related, except for the presence of the β-glucopyranose unit signals in 4. The glucose was confirmed to be located at C-4 from the HMBC correlation between H-1′ (δH 5.09) and C-4 (δC 159.4) (Fig. 5). D-Glucose were isolated from the enzymatic hydrolysate of 4 and identified by TLC and specific rotation. Therefore, the structure of 4 was elucidated as integriquinolone-4-O-β-D-glucopyranoside.
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δHa | δCb | δHa | δCb | δHa | δCb | |
| a In DMSO-d6 400 MHz for 1, 2, 3.b In DMSO-d6 100 MHz for 1, 2, 3. Coupling constants (J) in Hz are given in parentheses. The assignments were based on HSQC and HMBC experiments.c Signal overlapped. | ||||||
| 1 | 134.4 s | 134.6 s | 134.8 s | |||
| 2, 6 | 7.79, br d (7.2) | 127.1 d | 7.77, br d (7.2) | 127.1 d | 7.79, br d (7.6) | 127.6 d |
| 3, 5 | 7.44, t (7.6) | 128.3 d | 7.43, t (7.6) | 128.4 d | 7.46, t (7.6) | 128.8 d |
| 4 | 7.51, m | 131.1 d | 7.50, m | 131.0 d | 7.52, m | 131.6 d |
| 7 | 166.2 s | 166.4 s | 166.8 s | |||
| 8 | 3.69, m; 3.55, m | 44.5 t | 3.50, m | 45.6 t | 3.73, m; 3.57, m | 45.2 t |
| 9 | 4.83, t (4.0) | 78.8 d | 4.99, t (6.6) | 76.1 d | 4.90, m | 79.6 d |
| 10 | 132.5 s | 131.2 s | 141.1 s | |||
| 11, 15 | 7.32, br d (8.8) | 127.9 d | 7.36, br d (8.8) | 128.2 d | 7.41, br d (7.6) | 127.1 d |
| 12, 14 | 6.87, br d (8.8) | 113.3 d | 6.90, br d (8.8) | 113.5 d | 7.33, br d (7.4) | 128.4 d |
| 13 | 158.5 s | 158.7 s | 7.26, t (7.2) | 127.8 d | ||
| Glc-1′ | 4.42, d (7.6) | 102.9 d | 3.98, d (7.6) | 100.0 d | 4.46, d (7.6) | 103.5 d |
| Glc-2′ | 3.07,c m | 73.8 d | 3.04,c m | 73.5 d | 3.10, m | 74.3 d |
| Glc-3′ | 3.16, m | 76.5 d | 3.04,c m | 76.6 d | 3.18, m | 77.0 d |
| Glc-4′ | 3.07,c m | 69.9 d | 3.04,c m | 70.2 d | 3.07,c m | 70.4 d |
| Glc-5′ | 3.07,c m | 76.8 d | 2.96, m | 76.9 d | 3.08,c m | 77.4 d |
| Glc-6′a | 3.56, m | 60.9 t | 3.39, m | 61.2 d | 3.37, m | 61.4 t |
| Glc-6′b | 3.37, m | 3.65, m | 3.56, m | |||
| NH | 8.33, t (4.0) | 8.23, t (5.5) | 8.35, t (5.4) | |||
| OCH3 | 3.73, s | 55.0 q | 3.74, s | 55.0 q | ||
| Position | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δHa | δCb | δHa | δCb | δHc | δCd | |
| a In DMSO-d6 600 MHz for 4 and 5.b In DMSO-d6 150 MHz for 4 and 5.c In MeOH-d4 600 MHz for 6.d In MeOH-d4 150 MHz for 6. Coupling constants (J) in Hz are given in parentheses. The assignments were based on HSQC and HMBC experiments.e Signal overlapped. | ||||||
| 2 | 162.3 s | 163.3 s | 164.8 s | |||
| 3 | 6.19, s | 99.4 d | 6.02, s | 96.7 d | 105.1 s | |
| 4 | 159.7 s | 163.5 s | 159.2 s | |||
| 4a | 115.4 s | 116.4 s | 120.9 s | |||
| 5 | 8.04, dd (7.8, 1.8) | 123.2 d | 7.87, dd (7.8, 1.2) | 123.2 d | 7.95, dd (8.4, 1.2) | 117.0 d |
| 6 | 7.28, m | 121.4 d | 7.28, m | 122.9 d | 7.39, dd (8.4, 7.8) | 125.0 d |
| 7 | 7.66, m | 131.5 d | 7.64,e m | 132.2 d | 7.48, dd (7.8, 1.2) | 114.4 d |
| 8 | 7.52, br d (8.4) | 114.6 d | 7.62,e m | 116.3 d | 153.0 s | |
| 8a | 139.5 s | 139.5 s | 137.7 s | |||
| 9 | 3.57, 3H, s | 28.7 q | 5.87, d (9.6); 5.75, d (9.6) | 69.9 t | ||
| Glc-1′ | 5.09, d (7.8) | 99.5 d | 4.42, d (7.8) | 101.9 d | 5.04, d (7.8) | 103.0 d |
| Glc-2′ | 3.90, m | 73.0 d | 2.95, m | 73.9 d | 3.74, m | 74.7 d |
| Glc-3′ | 3.34, m | 76.1 d | 3.12,e m | 77.5 d | 3.59, m | 77.3 d |
| Glc-4′ | 3.19, m | 69.5 d | 3.07, m | 70.6 d | 3.47, m | 71.6 d |
| Glc-5′ | 3.46, m | 77.2 d | 3.10,e m | 77.9 d | 3.57, m | 78.5 d |
| Glc-6′ | 3.48, m; 3.70, m | 60.6 t | 3.47, m; 3.66, m | 61.7 t | 3.98, m; 3.75, m | 62.6 t |
| OCH3 | 3.96, s | 57.1 q | 4.52, s | 60.2 q | ||
| α | 7.86, d (2.4) | 145.4 d | ||||
| β | 7.40, d (2.4) | 106.5 d | ||||
 | ||
| Fig. 5 Key 1H, 1H-COSY and HMBC correlations of compounds 4, 5 and 6. | ||
Clausenaside E (5) was obtained as a white powder. Its HRESIMS displayed m/z [M + H]+ 368.1346 (calcd for C17H22NO8, 368.1340), which was consistent with a molecular formula of C17H21NO8 with eight indices of hydrogen deficiency. Compound 5 showed similar UV absorptions to those of 4, which suggested that their structure were closely related except that the 4-methoxy group and 9-hydroxymethyl-O-β-D-glucopyranoside of 5 replaced 4-O-β-D-glucopyranoside group and the 9-methyl group of 4 respectively. The linkage of the β-D-glucose moiety to C-9 was supported by the HMBC correlation observed between H-1′ (δH 4.42) and C-9 (δC 69.9) (Fig. 5). D-Glucose were isolated from the enzymatic hydrolysate of 5 and identified by TLC and specific rotation. Therefore, the structure of 5 was defined as 4-methoxy-1-hydroxymethyl-2(1H)-quinolinone-9-O-β-D-glucopyranoside.
Clausenaside F (6) was obtained as a white powder. Its molecular formula was assigned as C18H19NO8 on the basis of its 13C NMR and HRESIMS (m/z 378.1180 [M + H]+, calcd for C18H20NO8 378.1183; 400.0999 [M + Na]+, calcd for C18H19NO8Na, 400.1003), implying ten indices of hydrogen deficiency. The 1H and 13C NMR of 6 displayed signals characteristic of quinoline alkaloid glycoside, which were similar to these reported for robustine.15 The only difference was that the β-D-glucopyranose unit in 6 substituted the hydroxyl group in robustine. The D-glucose was localized at C-8, which was supported by the HMBC correlations (Fig. 5) from H-1′ (δH 5.04) to C-8 (δC 153.0). D-Glucose were isolated from the enzymatic hydrolysate of 6 and identified by TLC and specific rotation. Therefore, the structure of 6 was elucidated as robustine-8-O-β-D-glucopyranoside.
Clausenaside G (7) was obtained as a yellow powder. Its molecular formula was established as C15H17NO3 by the HRESIMS at m/z 282.1102 [M + Na]+, (calcd for C15H17NO3Na, 282.1101), indicating eight indices of hydrogen deficiency. The 1H and 13C NMR of 7 displayed signals characteristic of quinoline alkaloid, which was an analog of 3-hydroxyquinoline-2,4(1H,3H)-diones.16 In the HMBC spectrum (Fig. 6) of 7, correlations of H-1′ (δH 2.42)/C-2 (δC 171.9), C-3 (δC 82.0), C-4 (δC 195.1), C-2′ (δC 116.3), C-3′ (δC 135.2) and of H-4′ (δH 1.49) and H-5′ (δH 1.29)/C-2′ (δC 116.3), C-3′ (δC 135.2) suggested that the isopentene group was attached to C-2. The absolute configuration of 7 was defined as 3S by comparison of the experimental ECD spectra and calculated ECD data using the time-dependent density functional theory (TDDFT) method at the B3LYP/6-31G (d) level.17 The calculated ECD spectrum of 3S (7a) (Fig. 7) matched the experimental spectrum very well. Thus, the structure of 7 was elucidated.
 | ||
| Fig. 6 Key 1H, 1H-COSY and HMBC correlations of compounds 7 and 8. | ||
 | ||
| Fig. 7 Calculated ECD spectra of 7a (3S)- and 7b (3R)-isomers and the experimental ECD spectrum of 7. | ||
Clausenaside H (8) was obtained as a white powder. Its molecular formula C15H17NO3 was deduced from the HRESIMS (m/z 260.1280 [M + H]+, calcd for C15H18NO3, 260.1281; 282.1102 [M + Na]+, calcd for C15H17NO3Na 282.1101) and 13C NMR spectroscopic data, corresponding with eight indices of hydrogen deficiency. The 1H and 13C NMR of 8 displayed signals characteristic of quinoline alkaloid, which were similar to these reported for atanine.18 The only difference between 8 and atanine was that a methyl group in atanine is replaced by a hydroxymethyl group [δH 3.76 (2H, s, H-4′), δC 66.2] in 8. This was supported further by the HMBC correlations (Fig. 6) between H-4′ (δH 3.76) and C-2′ (δC 120.7)/C-3′ (δC 135.8)/C-5′ (δC 13.7). The NOE difference experiment displayed that a strong enhancement of H-2′ was observed when H-4′ was irradiated, while H-2′ was no enhanced on irradiation of H-5′, indicating an E configuration of the double bond. Thus, the structure of 8 was elucidated.
The ten known alkaloids were identified as 4-methoxy-8-O-β-D-glucopyranosyloxy-2(1H)-quinolinone (9),19 integriquinolone (10),20 araliopsine (11),21 clausenamide (12),22 4-methoxy-2(1H)-quinolone (13),23 4-methoxy-1-methyl-2-quinolone (14),24 neoclausenamide (15),22 demethylsecoclausenamide (16),25 ribalinine (17),21 claulansine D (18)5 based on the analysis and comparison with their spectroscopic data (NMR, UV and MS) with literature data.
Compounds 1–18 were tested for their inhibitory effects on LPS-induced NO production in murine microglial BV2 cells. In the anti-inflammatory assay, compounds 4, 6, 9, 17, 18 exhibited moderate inhibitory effect on LPS-stimulated NO production in murine microglial BV2 cells with curcumin as a positive control as shown in Table 4, whereas the other compounds were inactive in this assay (IC50 value > 10 μM).
| Position | 7 | 8 | ||
|---|---|---|---|---|
| δHa | δCb | δHa | δCb | |
| a In DMSO-d6 (600 MHz for 7, 400 MHz for 8).b In DMSO-d6 (150 MHz for 7, 100 MHz for 8). Coupling constants (J) in Hz are given in parentheses. The assignments were based on HSQC and HMBC experiments. | ||||
| 2 | 171.9 s | 163.3 s | ||
| 3 | 82.0 s | 121.9 s | ||
| 4 | 195.1 s | 160.7 s | ||
| 4a | 120.6 s | 116.0 s | ||
| 5 | 7.76, dd (7.6, 1.6) | 126.7 d | 7.69, dd (8.0, 1.0) | 122.5 d |
| 6 | 7.20, t (7.6) | 122.8 d | 7.20, td (8.0, 0.8) | 121.7 d |
| 7 | 7.70, td (7.0, 1.6) | 136.0 d | 7.49, td (8.0, 1.4) | 130.0 d |
| 8 | 7.33, d (8.3) | 115.5 d | 7.33, dd (8.0, 1.4) | 115.3 d |
| 8a | 142.6 s | 137.7 s | ||
| 1′ | 2.42, 2H, m | 39.4 t | 3.27, 2H, d (6.8) | 22.4 t |
| 2′ | 4.91, t (7.7) | 116.3 d | 5.40, td (6.8, 1.2) | 120.7 d |
| 3′ | 135.2 s | 135.8 s | ||
| 4′ | 1.49, s | 25.8 q | 3.76, 2H, s | 66.2 t |
| 5′ | 1.29, s | 17.4 q | 1.71, s | 13.7 q |
| 6′ | 171.9 s | |||
| NH | 11.72, br s | |||
| N-CH3 | 3.36, 3H, s | 29.7 q | ||
| OCH3 | 3.87, s | 61.5 q | ||
Experimental
Optical rotations were measured on a JASCO P2000 automatic digital polarimeter. UV spectra were recorded on a JASCO V-650 spectrophotometer, CD spectra were measured on a JASCO J-815 spectropolarimeter. IR spectra were recorded on a Nicolet 5700 spectrometer using an FT-IR microscope transmission method. NMR spectra were acquired with Bruker AVIIIHD 600, VNS-600, and Mercury-400 spectrometers in DMSO-d6 and MeOH-d4. HRESIMS spectra were collected on an Agilent 1100 series LC/MSD ion trap mass spectrometer. MPLC system was composed of two C-605 pumps (Büchi), a C-635 UV detector (Büchi), a C-660 fraction collector (Büchi), and an ODS column (450 mm × 60 mm, 50 μm, 400 g; YMC). Semi-preparative HPLC was conducted using a Shimadzu LC-6AD instrument with an SPD-20A detector and a Daicel Chiralpak AD-H column (250 × 10 mm, 5 μm). Preparative HPLC was also performed on a Shimadzu LC-6AD instrument with a YMC-Pack ODS-A column (250 × 20 mm, 5 μm). Column chromatography (CC) was performed with silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, People's Republic of China), SF-PRP 512A (100–200 mesh, Beijing Sunflower and Technology Development Co., Beijing, People's Republic of China), ODS (50 μm, YMC, Japan), and Sephadex LH-20 (GE, Sweden). TLC was carried out on glass precoated silica gel GF254 plates. Spots were visualized under UV light or by spraying with 10% sulfuric acid in EtOH followed by heating. Curcumin (99%, Sinopharm Chemical Reagent Company Limited).Plant materials
The stems of C. lansium were collected in Liuzhou, Guangxi, China, in March 2013 and identified by Engineer Guangri Long, Forestry of Liuzhou. A voucher specimen has been deposited at the Herbarium of Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College (ID-S-2320).Extraction and isolation
Air-dried, powdered stems of C. lansium (200 kg) were extracted with 95% ethanol (1000 L × 2 h × 3). The residue was suspended in water and then partitioned with EtOAc (3 × 40 L), and n-BuOH (3 × 40 L), successively. After removing the solvent, the n-BuOH-soluble portion (850 g) was fractionated via macroporous adsorbent resin (HPD-100) column with H2O, 30% EtOH, 60% EtOH, and 95% EtOH to yield four corresponding fractions A–D.Fraction B (180 g) was fractionated via silica gel column chromatography, eluting with CHCl3–MeOH–H2O (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.05, 9:1:0.1, 8:2:0.2, 7:3:0.3, 6:4:0.4) to afford twelve fractions B1-B12 on the basis of TLC analysis. Fraction B4 (5.9 g) was further separated by reversed-phase silica MPLC with 20%–50% MeOH (50 mL min−1, 6 h) to afford B4-1-B4-47 fractions. Fractions B4-38-B4-42 were successively separated using preparative HPLC (detection at 210 nm, 18% CH3CN, 8 mL min−1) to yield 1 (29 mg, tR 83.45 min), 2 (5.0 mg, tR 73.87 min), 3 (19 mg, tR 67.02 min) and 6 (9 mg, tR 78.25 min). Fraction B6 (8.3 g) was further separated by reversed-phase silica MPLC with 20%–45% MeOH (50 mL min−1, 6 h) to afford fractions B6-1-B6-39. Fraction B6-31-B6-32 was successively separated using preparative HPLC (detection at 210 nm, 15% CH3CN, 8 mL min−1) to yield 5 (3 mg, tR 51.14 min). Fraction B7 (36.8 g) was fractionated via silica gel column chromatography, eluting with CHCl3–MeOH–H2O (10:1:0.5–8:2:0.3) to afford fractions B7-1-B7-18 on the basis of TLC analysis. Fraction B7-6 (11.7 g) was further separated by reversed-phase silica MPLC with 20%–50% MeOH (50 mL min−1, 8 h) to afford B7-6-1-B7-6-11 fractions. Fraction B7-6-5 was successively separated using preparative HPLC (detection at 210 nm, 12% CH3CN, 8 mL min−1) to yield 4 (16 mg, tR 40.59 min). Fraction B7-6-8 was successively separated using preparative HPLC (detection at 210 nm, 11% CH3CN, 8 mL min−1) to yield 9 (16 mg, tR 40.59 min).
:1:0.05, 9:1:0.1, 8:2:0.2, 7:3:0.3, 6:4:0.4) to afford twelve fractions B1-B12 on the basis of TLC analysis. Fraction B4 (5.9 g) was further separated by reversed-phase silica MPLC with 20%–50% MeOH (50 mL min−1, 6 h) to afford B4-1-B4-47 fractions. Fractions B4-38-B4-42 were successively separated using preparative HPLC (detection at 210 nm, 18% CH3CN, 8 mL min−1) to yield 1 (29 mg, tR 83.45 min), 2 (5.0 mg, tR 73.87 min), 3 (19 mg, tR 67.02 min) and 6 (9 mg, tR 78.25 min). Fraction B6 (8.3 g) was further separated by reversed-phase silica MPLC with 20%–45% MeOH (50 mL min−1, 6 h) to afford fractions B6-1-B6-39. Fraction B6-31-B6-32 was successively separated using preparative HPLC (detection at 210 nm, 15% CH3CN, 8 mL min−1) to yield 5 (3 mg, tR 51.14 min). Fraction B7 (36.8 g) was fractionated via silica gel column chromatography, eluting with CHCl3–MeOH–H2O (10:1:0.5–8:2:0.3) to afford fractions B7-1-B7-18 on the basis of TLC analysis. Fraction B7-6 (11.7 g) was further separated by reversed-phase silica MPLC with 20%–50% MeOH (50 mL min−1, 8 h) to afford B7-6-1-B7-6-11 fractions. Fraction B7-6-5 was successively separated using preparative HPLC (detection at 210 nm, 12% CH3CN, 8 mL min−1) to yield 4 (16 mg, tR 40.59 min). Fraction B7-6-8 was successively separated using preparative HPLC (detection at 210 nm, 11% CH3CN, 8 mL min−1) to yield 9 (16 mg, tR 40.59 min).
Fraction C (220 g) was partitioned via kieselguhr eluting with CHCl3, EtOAc, n-BuOH, acetone, and MeOH to afford five fractions C1-C5. After the solvent was removed, the C1 fraction (43 g) was subjected to SF-PRP 512A resin eluting with 35%–70% MeOH to afford fractions C1-1-C1-6. C1-2 (7.1 g) was further separated by reversed-phase silica MPLC with 30%–50% MeOH (50 mL min−1, 6 h) to afford fractions C1-2-1-C1-2-11. Fraction C1-2-4 was subjected to Sephadex LH-20 and preparative HPLC (detection at 210 nm, 19% CH3CN, 8 mL min−1) to yield 10 (8 mg, tR 39.04 min). Fraction C1-2-5 was subjected to Sephadex LH-20 and preparative HPLC (detection at 210 nm, 19% CH3CN, 8 mL min−1) to yield 11 (7 mg, tR 32.04 min). Fraction C1-2-8 was subjected to Sephadex LH-20 and preparative HPLC (detection at 210 nm, 25% CH3CN, 8 mL min−1) to yield 12 (153 mg, tR 21.99 min) and 13 (8 mg, tR 30.68 min). Fraction C2 (14.6 g) was further separated by reversed-phase silica MPLC with 35%–55% MeOH (50 mL min−1, 10 h) to afford fractions C2-1-C2-15. Fractions C2-11 was subjected to Sephadex LH-20 and preparative HPLC (detection at 210 nm, 29% CH3CN, 8 mL min−1) to yield 7 (2 mg, tR 46.59 min), 14 (80 mg, tR 38.00 min) and 15 (54 mg, tR 38.20 min). Fractions C2-12 was subjected to Sephadex LH-20 and preparative HPLC (detection at 210 nm, 30% CH3CN, 8 mL min−1) to yield 8 (10 mg, tR 28.20 min), 16 (84 mg, tR 45.00 min), 17 (40 mg, tR 41.81 min) and 18 (4 mg, tR 42.87 min).
Structure characterization
ε) 201.6 (4.31), 225.6 (4.14) nm; IR (microscope) νmax 3314, 2919, 1640, 1617, 1540, 1515, 1246, 1103, 1075, 1036, 828, 697 cm−1; CD (MeOH) λmax (Δε) 217.5 (+2.28), 238 (−2.86) nm; 1H NMR (DMSO-d6, 400 MHz) and 13C NMR (DMSO-d6, 100 MHz), see Table 1; HRESIMS m/z 456.1634 [M + Na]+ (calcd for C22H27NO8Na, 456.1629).ε) 201.8 (4.61), 226.2 (4.38) nm; IR (microscope) νmax 3361, 2924, 1641, 1613, 1577, 1543, 1513, 1306, 1248, 1077, 1029, 833, 713 cm−1; CD (MeOH) λmax (Δε) 218 (−1.48), 237 (+2.71) nm; 1H NMR (DMSO-d6, 400 MHz) and 13C NMR (DMSO-d6, 100 MHz), see Table 1; HRESIMS m/z 456.1635 [M + Na]+ (calcd for C22H27NO8Na, 456.1629).ε) 201.6 (4.14) nm; IR (microscope) νmax 3357, 2917, 1641, 1577, 1541, 1490, 1313, 1159, 1076, 701 cm−1; CD (MeOH) λmax (Δε) 203.5 (+2.94), 228 (−1.90) nm; 1H NMR (DMSO-d6, 400 MHz) and 13C NMR (DMSO-d6, 100 MHz), see Table 1; HRESIMS at m/z 426.1524 [M + Na]+ (calcd for C21H25NO7Na, 426.1523).ε) 228.2 (4.39), 269.6 (3.48), 320.4 (3.34) nm; IR (microscope) νmax 3501, 3362, 3273, 2930, 2882, 1640, 1615, 1574, 1109, 1080, 841, 755 cm−1; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz), see Table 2; HRESIMS m/z 338.1232 [M + H]+ (calcd for C16H20NO7, 338.1234).ε) 226.6 (4.38), 268 (3.50), 314.6 (3.22) nm; IR (microscope) νmax 3391, 2923, 1647, 1581, 1463, 1400, 1326, 1245, 1113, 1071, 828, 775, 759 cm−1; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz), see Table 2; HRESIMS m/z 368.1346 [M + H]+ (calcd for C17H22NO8, 368.1340), 390.1161 [M + Na]+ (calcd for C17H21NO8Na, 390.1159).ε) 202.4 (4.24), 243.2 (4.53) nm; IR (microscope) νmax 3342, 2935, 1589, 1516, 1460, 1397, 1373, 1267, 1094, 1034, 748 cm−1; 1H NMR (MeOH-d4, 600 MHz) and 13C NMR (MeOH-d4, 150 MHz), see Table 2; HRESIMS m/z 378.1180 [M + H]+ (calcd for C18H20NO8, 378.1183), 400.0999 [M + Na]+ (calcd for C18H19NO8Na, 400.1003).ε) 201.8 (4.13), 234.8 (4.27) nm; IR (microscope) νmax 3394, 2920, 2849, 1709, 1663, 1603, 1472, 1368, 1302, 1123, 1102, 759 cm−1; 1H NMR (DMSO-d6, 600 MHz) and 13C NMR (DMSO-d6, 150 MHz), see Table 3; HRESIMS m/z 282.1102 [M + Na]+ (calcd for C15H17NO3Na, 282.1101).ε) 226.6 (4.41), 270.6 (3.74), 322.8 (3.71) nm; IR (microscope) νmax 3295, 3174, 2997, 2945, 2898, 2853, 1659, 1612, 1571, 1501, 1434, 1379, 1267, 1094, 1001, 751 cm−1; 1H NMR (DMSO-d6, 400 MHz) and 13C NMR (DMSO-d6, 100 MHz), see Table 3; HRESIMS m/z 260.1280 [M + H]+ (calcd for C15H18NO3, 260.1281), 282.1102 [M + Na]+ (calcd for C15H17NO3Na, 282.1101).Enzymatic hydrolysis of 1–6
Compounds 1–6 (1.0–8.0 mg) were hydrolyzed in H2O (1 mL) with snailase at 37 °C for 4 h, then extracted by EtOAC (3 × 3 mL). The aqueous phase was dried by a stream of N2, then chromatographed over silica gel eluting with CH3CN–H2O (8:1), to yield glucose. The glucose (0.3–1.2 mg) from the hydrolysates of 1–6 gave retention factor (Rf ≈ 0.33; CH3CN–H2O, 6:1) on TLC, [α]25D values +43.1 to +68.6 (c = 0.02–0.1, H2O)], and 1H NMR spectral data (D2O) consistent with those of an authentic D-glucose. The EtOAC phase was concentrated under reduced pressure, and purified by RP-HPLC to obtain 1a (3.14 mg) from 1. (S)-(+)-tembamide (1a): white amorphous powder; [α]25D +54.8 (c 0.2 CHCl3); CD (MeOH) λmax (Δε) 216 (+0.21), 235 (−2.14) nm; 1H NMR (DMSO-d6, 400 MHz) δH 8.46 (NH, t, J = 6.0 Hz), 7.83 (2H, d, J = 7.6 Hz), 7.50 (1H, m), 7.44 (2H, m), 7.28 (2H, d, J = 8.0 Hz), 6.89 (2H, d, J = 8.0 Hz), 5.41 (1H, d, J = 4.0 Hz), 4.72 (1H, m), 3.72 (3H, s), 3.43 (1H, m), 3.28 (1H, m); 13C NMR (DMSO-d6, 150 MHz) δC 166.3, 158.3, 135.8, 134.5, 131.0, 128.2, 128.2, 127.2, 127.2, 127.1, 127.1, 113.4, 113.4, 70.7, 55.0, 47.7; HRESIMS at m/z 294.1109 [M + Na]+ (calcd for C16H17NO3Na, 294.1101).
Preparation of (R)- and (S)-MTPA esters of 1a
A solution of 1a (1.09 mg) in dehydrated CH2Cl2 (2 mL) was treated with (S)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride [(S)-MTPA-Cl (10 mg)] in the presence of anhydrous pyridine, and the mixture was stirred at room temperature for 13 h. After cooling, the reaction mixture was poured into ice-water and extracted with EtOAC. The EtOAC extract was successively washed with 5% aqueous HCl, saturated aqueous NaHCO3, and brine, then dried over Na2SO4 and filtered. The solvent was removed from the filtrate under reduced pressure to afford a residue.26 The residue was purified by semi-preparative HPLC (C18 column, 3.0 mL min−1, UV 210 nm, 58% CH3CN–H2O) to yield (S)-MTPA ester derivative of 1a (compound 1aa 1.12 mg). Using a similar procedure, (R)-MTPA ester derivative of 1a (compound 1ab 0.93 mg) was obtained from 1a (1.04 mg).Inhibition of nitric production assay
The murine microglial BV2 cells, purchased from the Cell Culture Centre at the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), at 37 °C atmosphere, 5% CO2, and 100% relative humidity. The microglia cells were placed in 96-well cell culture plates (2 × 104 cell per mL) and preincubated for 24 h. Then the cells were treated with various concentrations of isolated compounds in triplicate for 1 h and continuously incubated with LPS (Sigma-Aldrich) (0.3 μg mL−1) for 24 h. Curcumin was used as the positive control. After incubation, the supernatants (100 μL) were added to a solution of 100 μL of Griess reagent (a 1:1 mixture of 0.1% naphthylethylenediamine and 1% sulfanilamide in 5% H3PO4) at room temperature for 20 min. NO concentration was quantified by a microplate reader at 540 nm for the amount of stable nitrite produced in the cell culture supernatants using the Griess assay.27
Acknowledgements
We are grateful to the Department of Instrumental Analysis, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, for the measurement of the UV, IR, CD, NMR, and HRESIMS spectra. This research program is financially supported by the National Natural Science Foundation of China (No. 21272278) and the National Megaproject for Innovative Drugs (No. 2012ZX09301002-002).Notes and references
- A. C. Adebajo, E. O. Iwalewa, E. M. Obuotor, G. F. Ibikunle, N. O. Omisore, C. O. Adewunmi, O. O. Obaparusi, M. Klaes, G. E. Adetogun, T. J. Schmidt and E. J. Verspohl, J. Ethnopharmacol., 2009, 122, 10–19 CrossRef CAS PubMed.
- W. Maneerat, T. Ritthiwigrom, S. Cheenpracha and S. Laphookhieo, Phytochem. Lett., 2012, 5, 26–28 CrossRef CAS PubMed.
- G. T. Liu, W. X. Li, Y. Y. Chen and H. L. Wei, Drug Dev. Res., 1996, 39, 174–178 CrossRef CAS.
- K. N. Prasad, H. H. Xie, J. Hao, B. Yang, S. X. Qiu, X. Y. Wei, F. Chen and Y. M. Yue, Food Chem., 2010, 118, 62–66 CrossRef CAS PubMed.
- H. Liu, C. J. Li, J. Z. Yang, N. Ning, Y. K. Si, L. Li, N. H. Chen, Q. Zhao and D. M. Zhang, J. Nat. Prod., 2012, 75, 677–682 CrossRef CAS PubMed.
- H. Liu, F. Li, C. J. Li, J. Z. Yang, L. Li, N. H. Chen and D. M. Zhang, Phytochemistry, 2014, 107, 141–147 CrossRef CAS PubMed.
- Y. Q. Du, H. Liu, C. J. Li, J. Ma, D. Zhang, L. Li, H. Sun, X. Q. Bao and D. M. Zhang, Fitoterapia, 2015, 103, 122–128 CrossRef CAS PubMed.
- Q. Zhao, J. Z. Yang, C. J. Li, N. H. Chen and D. M. Zhang, J. Asian Nat. Prod. Res., 2011, 13, 361–366 CrossRef CAS PubMed.
- A. Shoeb, R. S. Kapil and S. P. Popli, Phytochemistry, 1973, 12, 2071–2072 CrossRef CAS.
- A. Kamal, A. A. Shaik, M. Sandbhor and M. S. Malik, Tetrahedron: Asymmetry, 2004, 15, 3939–3944 CrossRef CAS PubMed.
- M. Yoshikawa, T. Morikawa, Y. Zhang, S. Nakamura, O. Muraoka and H. Matsuda, J. Nat. Prod., 2007, 70, 575–583 CrossRef CAS PubMed.
- J. M. Seco, E. Quinoa and R. Riguera, Chem. Rev., 2004, 104, 17–117 CrossRef CAS.
- K. H. Kim, Y. J. Park, K. H. Chuang, M. L. R. Yip, J. Clardy, D. Senger and S. Cao, J. Nat. Prod., 2015, 78, 320–324 CrossRef CAS PubMed.
- D. Lai, H. B. Oesterhelt, W. E. G. Muller, V. Wray and P. Proksch, Fitoterapia, 2013, 91, 100–106 CrossRef CAS PubMed.
- I. M. Fakhrutdinova, G. P. Sidyakin and S. Y. Yunusov, Khim. Prir. Soedin., 1965, 1, 107–109 Search PubMed.
- S. Kafka, K. Proisl, V. Kasparkova, D. Urankar, R. Kimmel and J. Kosmrlj, Tetrahedron, 2013, 69, 10826–10835 CrossRef CAS PubMed.
- C. Wang, C. J. Li, J. Ma, J. Z. Yang, X. G. Chen, L. Li and D. M. Zhang, RSC Adv., 2015, 5, 30046 RSC.
- S. Perrett and P. J. Whitfield, Planta Med., 1995, 61, 276–278 CrossRef CAS PubMed.
- Y. F. Su, Y. Luo, C. Y. Guo and D. A. Guo, J. Asian Nat. Prod. Res., 2004, 6, 223–227 CrossRef CAS PubMed.
- H. Ishii, K. Koyama, I. S. Chen, S. T. Lu and T. Ishikawa, Chem. Pharm. Bull., 1982, 30, 1992–1997 CrossRef CAS.
- N. M. D. Brown, M. F. Grundon, D. M. Harrison and S. A. Surgenor, Tetrahedron, 1980, 36, 3579–3584 CrossRef CAS.
- M. H. Yang, Y. Y. Chen and L. Huang, Phytochemistry, 1988, 27, 445–450 CrossRef CAS.
- C. Kaneko, T. Suzuki, M. Sato and T. Naito, Chem. Pharm. Bull., 1987, 35, 112–123 CrossRef CAS.
- M. N. S. Nayar, C. V. Sutar and M. K. Bhan, Phytochemistry, 1971, 10, 2843–2844 CrossRef CAS.
- M. H. Yang and L. Huang, Chin. Chem. Lett., 1991, 2, 775–776 CAS.
- L. Ni, X. Zhou, J. Ma, X. M. Zhang, C. J. Li, L. Li, D. J. Yang, Y. Y. Shao, S. B. Zhou, T. T. Zhang and D. M. Zhang, J. Asian Nat. Prod. Res., 2015, 17, 615–624 CrossRef CAS PubMed.
- D. Zhang, R. Liu, L. Sun, C. Huang, C. Wang, D. M. Zhang, T. T. Zhang and G. H. Du, Molecules, 2011, 16, 3875–3884 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The spectra including 1D-, 2D-NMR, HRESIMS of compounds 1–8 as well as related original CD spectrum for Clausenasides A (1), B (2) and C (3), and ECD calculation data for Clausenaside G (7). See DOI: 10.1039/c5ra14173g |
| This journal is © The Royal Society of Chemistry 2015 |