
Formation of polysaccharide aerogels in ethanol
Gabrijela Tkalec,
Željko Knez and
Zoran Novak*
University of Maribor, Faculty of Chemistry and Chemical Engineering, Smetanova 17, SI-2000, Maribor, Slovenia. E-mail: zoran.novak@um.si; Fax: +386 2 2527 774; Tel: +386 2 2294 405
First published on 8th September 2015
Abstract
Aerogels are outstanding materials, obtained by the sol–gel process. The production of polysaccharide aerogels is however time-consuming and their use for life-science applications is limited. To accelerate the production time, ethanol was used to induce the gelation of pectin, alginate, xanthan and guar gum. Polysaccharide aerogels were produced by dissolution in water, gelation in ethanol and supercritical drying. Only ethanol was used for the gelation without the use of any other cross-linking agent. In addition there was no solvent-exchange step prior to supercritical drying since the gelation occurred directly in ethanol. Differential scanning calorimetry was used to analyze the decompositions of the samples and also to measure their thermal conductivities. SEM and rheological analyses were performed in order to characterize the new materials. The prepared dry materials were highly porous and possessed some outstanding properties, namely, the low-methoxyl and high-methoxyl pectin monolithic aerogels possessed the highest surface areas yet reported, 510 m2 g−1 and 390 m2 g−1, respectively. To our knowledge this is the first known report regarding synthesis and characterization of pure xanthan and guar aerogels. Their surface areas were also high, 370 m2 g−1 and 110 m2 g−1, respectively. Very low thermal conductivity was observed for pectin aerogels, 0.021 W m−1 K−1.
1. Introduction
Polysaccharides are materials, found in abundance in nature. They possess many interesting properties and due to their relatively low cost they have been widely used in various applications.1–3 This research work is focused on low-methoxyl (LM) pectin, high-methoxyl (HM) pectin, alginate, guar and xanthan gum (Fig. 1), which are nowadays mainly used in the food or pharmaceutical industry due to their gelling and stabilizing abilities.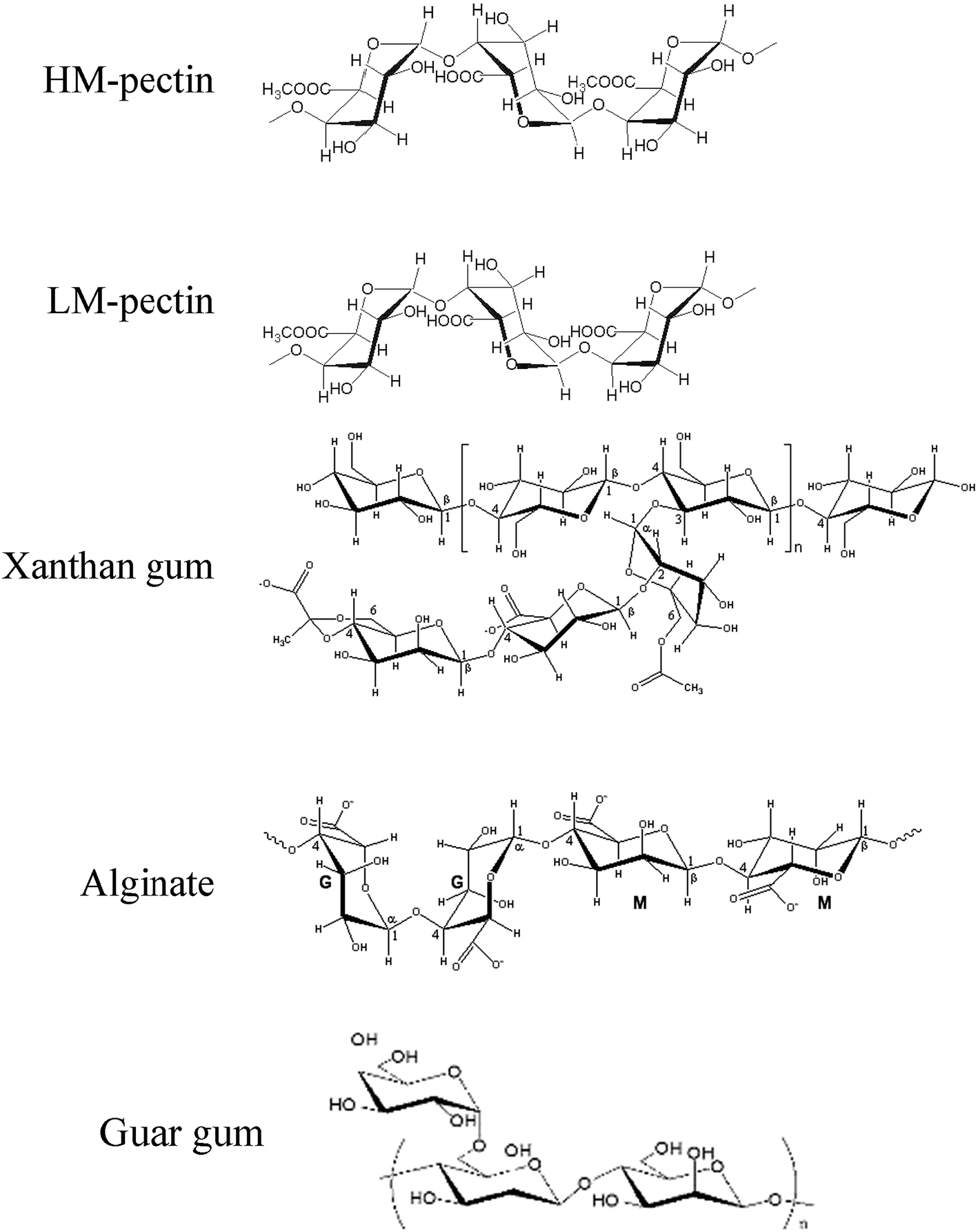 | ||
| Fig. 1 Structural formula of polysaccharides, used in this research work. | ||
All the above-mentioned polysaccharides could be used during the sol–gel process for obtaining wet gels. By supercritical technology they could be converted into highly sophisticated materials – aerogels. LM-pectin aerogels were until now prepared mostly by cross-linking with calcium or other divalent ions4,5 and HM-pectin aerogels by thermal or acidic gelation.6–9 Alginate aerogels have been highly researched materials in the recent past.10–12 The gelation of this naturally-occurring polysaccharide is obtained by divalent or multivalent cations (except Mg2+).13 Xanthan gum/clay aerogels were prepared by Wang et al.14 however, pure xanthan or guar aerogels have never been prepared as yet. Recently, the gelation of xanthan gum was performed in glycerol,15 but only wet gels were produced in this research. Pure guar, freeze-dried aerogels were prepared by enzymatic oxidation and possessed some outstanding properties, such as high compressive modulus.16 Table 1 briefly summarizes the already prepared polysaccharide monolithic aerogels together with their specific surface areas.
| Aerogel | Crosslinking method | Polysaccharide concentration (%) | SBET (m2 g−1) | Reference |
|---|---|---|---|---|
| HM-pectin | Acid gelation | 5 | 200 | 9 |
| HM-pectin | Thermal gelation | 6 | 247 | 6 |
| HM-pectin | Acid gelation | 2–6 | 230–270 | 7 |
| LM-pectin | Calcium ions | 2 | 213 (apple) | 4 |
| 2 | 248 (citrus) | |||
| Alginate | Calcium ions | 2 | 150–300 | 17 |
| Alginate | Acid gelation | 2 | 391 | 18 |
| Alginate | Cation ions | 1 | 298–374 | 18 |
| Alginate | Cation ions | 1 | 187–356 | 19 |
| Alginate | Cation ions | 0.25–2.98 | 545–264 | 20 |
| Starch | Thermal gelation | 2 | 72 (potato) | 17 |
| 90 (corn) | ||||
| Cellulose | NaOH | 1 | 200 | 21 |
| Cellulose | NaOH | 5–7 | 200–240 | 22 |
| Cellulose | Ionic liquids | 3 | 230 | 23 |
| 15 | 130 | |||
| Cellulose | Catalyst 2,2,6,6-tetramethylpiperidine-1-oxyl | 1 | 500–600 | 24 |
| Chitin | Acid gelation | 1 | 560 | 25 |
Aerogels are nowadays highly investigated by researchers due to their outstanding properties. Namely, they could be used as thermal insulators,26 as capacitors,27,28 and also within pharmaceutical or biomedical applications.29,30 Silica aerogels were highly investigated for their insulating properties and are nowadays classified as best insulating materials. However, silica aerogels can possess certain limitations, mostly due to their fragility. Mostly inorganic aerogels are supercritically dried to aerogel powders or highly cracked and fragile monoliths. Polysaccharide aerogels usually don't have those limitations and their production is straightforward. Cellulose aerogels were investigated for their thermal properties. Composites of cellulose and silica have been prepared in order to lower the thermal conductivity of pure cellulose and increase the mechanical properties of pure silica aerogel. The thermal conductivity of pure cellulose aerogel was 0.033 W m−1 K−1. In composite cellulose–silica aerogels, superinsulating silica aerogel is filling aerocellulose pores in general and macropores in particular: thermal conductivity is thus decreased to 0.027 ± 0.001 W m−1 K−1.31 Hayase et al. observed very low thermal conductivity for composite polymethylsilsesquioxane–cellulose aerogels as low as 0.015 W m−1 K−1. Rudaz et al.7 was the first to report the ultralow thermal conductivities of pectin in the range of 0.016–0.022 W m−1 K−1.
Anyhow, the production time of pectin or alginate aerogels is usually long, mostly due to the solvent exchange step which is needed prior to supercritical drying. Also, the sol–gel process sometimes requires longer time, depending on the gelation method. The overall production time of polysaccharide aerogels could thus be measured in days.9 In some cases the use of toxic compounds e.g. glutaraldehyde is also required for the gelation.
In this behalf, novel gelation methods based on natural materials are highly desirable. Therefore during this research we investigated a route to stable polysaccharide gels. Back in 1984 Oakenfull et al.32 proved that the addition of ethanol strengthens hydrophobic interactions between methoxyl groups in HM-pectins. Those results were essential for the research presented in this article. The theory on hydrophobic interactions was employed to investigate the gelations of selected polysaccharides in the presence of alcohol.
2. Experimental section
2.1. Materials
High-methoxyl (Pectin Classic CU-L 069/13, degree of esterification: 78%) and low-methoxyl pectin (Pectin amid AF 020, degree of esterification: 27–32%, degree of amidation: 18–23%) were kindly provided by Herbstraith&Fox, Germany. Alginic acid sodium salt (from brown algae, viscosity 100–300 cP), xanthan gum (viscosity 800–1200 cP) and guar were purchased from Sigma & Aldrich. Ethanol (absolute) was obtained from Sigma & Aldrich. CO2 (Messer) was used for supercritical drying.2.2. Methods
 | ||
| Fig. 2 Diffusion governed ethanol-induced gelation. | ||
To obtain dry materials from wet gels in order to measure their properties, supercritical drying was performed using CO2 at 40 °C and 150 bar. Other drying methods do exist, i.e. evaporation or lyophilization. However, it has been observed34,35 that supercritical drying is the best method for preserving the initial structure of the wet gel. First, the autoclave was filled with absolute ethanol and prepared wet gels were added thereafter. The temperature was increased to 40 °C and then the pressure was slowly increased up to 150 bar. The drying process for monoliths was performed for 6 h at the 200–300 L h−1 flow-rate. Then the system was slowly depressurized and left to cool down.
 | (1) |
The surface area, pore sizes and pore volumes of the polysaccharide aerogels were studied using low temperature nitrogen adsorption/desorption analysis. Prior to the measurements all the samples had been degassed under reduced pressure (<1 mPa) at 70 °C for 10 h. The specific surface area was then determined by the BET method. Pore volume was determined by filling the pore completely with liquid nitrogen at P/P0 = 0.99 and average pore size distribution was determined by the BJH adsorption method.
Scanning electron micrographs of the prepared aerogel samples were obtained using a Sirion 400NC scanning electron microscope (SEM). The samples were fractioned and then sputter-coated with gold particles and scanned at an accelerating voltage of 5–10 kV.
Thermal transitions were studied by a differential scanning calorimeter (DSC) with 5 °C min−1 heating-rate. DSC analysis was performed for studying the phase transitions like melting, glass transitions or exothermic decompositions. The analysis was performed on a TGA/DSC1 Mettler Toledo apparatus. The temperature range during analysis was set at between 30 °C to 600 °C.
The thermal conductivity was measured by a HP DSC1 Mettler Toledo. The method for measuring thermal conductivity using DSC was already described.36–39 In this research the method was as follows. Indium was used as the reference metal. About 80 mg of the metal was placed in a 40 μL light aluminum crucible without a lid. Aerogel samples were prepared as disks at heights of 0.5 to 1.5 mm. The surface of the aerogel was polished. The diameter of the samples was the same as that of the bottom of the crucible (6 mm). The sample was then analyzed from 130 to 160 °C at 0.5 K min−1. Nitrogen was used as a purge gas (50 mL min−1). The thermal conductivity was then calculated from eqn (2):
 | (2) |
Rheological measurements were performed at Anton Paar Ljubljana on MCR302 Rheometer. 1–2 mm thick wet gels were measured by the amplitude sweep (AS) method. The temperature at all measurements was 25 °C.
3. Results
3.1. Influence of polysaccharide on aerogel's characteristics
HM-pectin, LM-pectin, xanthan, alginate and guar aerogels were prepared from 4% polymer solutions. Macroscopically all samples appear monolithic and homogeneous as shown in Fig. 3. Alginate, xanthan gum and guar produced white wet gels, LM-pectin yellow and HM-pectin transparent. The term ‘alcogel’ is used when the solvent in the gel is alcohol. In contrast, the hydrogels are wet gels having water in the pores and aerogels are dry materials the pores of which are filled completely with air. | ||
| Fig. 3 Physical appearance of different polysaccharide monolithic gels before (alcogels) and after (aerogels) supercritical drying. | ||
It is well-known that junction zones are strengthened by hydrophobic interactions between methyl ester groups,32 which are needed for overcoming the entropic barrier to gelation. It was also proven that the addition of ethanol strengthens hydrophobic interactions between high-methoxyl pectin chains.32 The ethanol-induced gelation of polysaccharides is, to the best of our knowledge, a new method for producing stable monoliths from pectin, xanthan, alginate and guar. Gelation most probably occurs due to the low water activity. As aqueous polysaccharide solution comes in contact with the ethanol, polysaccharide–water interactions are minimized. Hydrophobic interactions between polysaccharide chains are increased so the gelation can occur. The model is presented in Fig. 4.
 | ||
| Fig. 4 Ethanol-induced gelation mechanism: (A) initial aqueous polysaccharide solution. (B) Ethanol increases hydrophobic interactions and induces the gelation. (C) Alcogel formation. | ||
The preparation method for the polysaccharide aerogels using the novel approach of ethanol-induced gelation was rather simple. Only a low amount of polysaccharide, distilled water and absolute ethanol were used for the wet gel preparation. Afterwards, ethanol and CO2 were used for obtaining stable dry materials, suitable for various applications. By this method, differently shaped monoliths could be obtained, ranging from thin membranes to high cylinders. Gelation step was performed in 1 h for disks of 15 mm diameter and 3 mm height and immediately afterwards the supercritical drying was performed for additional 6 h. Sometimes the presence of toxic compound is needed in order to obtain stable cross-linked gel (e.g. glutaraldehyde or glyoxal). In this research the final materials, obtained after ethanol-induced gelation contained no other substance as pure cross-linked polysaccharide. If immersed into the phosphate buffer solution, simulated gastric fluid or water, all prepared materials started to swell and eventually degrade.
The specific surface areas SBET were obtained with the BET method for all prepared aerogels. In general, all bioaerogels have significantly lower specific surface area as compared to silica aerogels (800–1000 m2 g−1). SBET varies from 110 m2 g−1 for guar and 510 m2 g−1 for LM-pectin. This is the first known report on specific surface area of guar aerogels, prepared after supercritical drying. In this research we also obtained HM- and LM-pectin monolithic aerogels with highest surface area ever reported. Higher surface areas were obtained only in the case of LM-pectin microspheres.4 As shown in Fig. 5, all isotherms are type IV (hysteresis), which is typical for mesoporous materials. Following the IUPAC classification, there are four types of hysteresis (H1, H2, H3 and H4). Isotherms in the Fig. 5 could be classified as H1, which is associated with porous materials, exhibiting a narrow distribution of relatively uniform (cylindrical-like) pores. Also, a high quantity of adsorbed gas is observed by HM-pectin, LM-pectin and xanthan gum aerogels.
 | ||
| Fig. 5 Adsorption/desorption isotherms for polysaccharide aerogels. | ||
Moreover, the obtained pore-sizes were within the range of 14–20 nm. The data on specific surface areas are collated within the Table 2. It is shown that both HM-pectin and LM-pectin monolithic aerogels possess high surface areas. The surface areas are at least twice higher than that reported in the literature for pectin monolithic aerogels. Therefore the ethanol-induced gelation seems a promising method for preparing pectin aerogels.
| Sample | Bulk densitya (g cm−3) | True densitya (g cm−3) | Porositya (%) | SBET (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) | Thermal conductivitya (W m−1 K−1) |
|---|---|---|---|---|---|---|---|
| a Average of five measurements. | |||||||
| HM-pectin | 0.0861 ± 0.0022 | 2.5805 ± 0.0551 | 96.6 ± 0.01 | 384 | 1.84 | 19 | 0.023 ± 0.002 |
| LM-pectin | 0.0771 ± 0.0008 | 2.5700 ± 0.0523 | 97.0 ± 0.03 | 510 | 1.87 | 17 | 0.021 ± 0.002 |
| Xanthan | 0.0961 ± 0.0021 | 1.8742 ± 0.0150 | 94.9 ± 0.01 | 363 | 1.50 | 20 | 0.028 ± 0.002 |
| Alginate | 0.1569 ± 0.0061 | 3.1804 ± 0.0325 | 95.0 ± 0.16 | 147 | 0.34 | 14 | 0.080 ± 0.003 |
| Guar | 0.2942 ± 0.0065 | 2.2451 ± 0.0233 | 86.9 ± 0.16 | 111 | 0.38 | 15 | 0.095 ± 0.003 |
To the best of our knowledge there is no research on the pure xanthan gum aerogels (obtained after supercritical drying) preparation in the literature. Therefore the comparison of surface areas is not possible. Pure wet xanthan gels could only be prepared by Cu(III) ions.40 In addition, some composites were prepared41,42 but the surface areas of those materials were much lower than obtained during this research work. Oppositely, the surface area of the alginate aerogels was only around 150 m2 g−1. The obtained surface area was in agreement of previously published research, however once higher surface areas had already been obtained by alginate monolithic aerogels.17 It was observed that by increasing the initial viscosity of the alginic acid, the shrinkage of the gel was lowered.43 It is expected that the shrinkage would impact on the final surface areas of the aerogels. Therefore this phenomena should be further investigated in order to maximize the surface areas of the alginate aerogels. Low bulk densities and high porosities were observed by all aerogels. LM-pectin once again provided the best results, having the lowest bulk density of 0.0771 g cm−3 and porosity of 97%.
SEM images of all prepared samples are shown in Fig. 6. Even though the same gelation method was employed for all the materials, the obtained aerogels are completely different within the structure. The difference in the aerogel structure is then without doubt the result of a polysaccharide. The evidence of macro-pores in aerogels is shown in the Fig. 6. Unfortunately we could not observed the meso or microporosity at those magnitudes. As seen from the Table 2, the average pore size in prepared aerogels is less than 20 nm. This could not be seen on SEM figures, due to the lower magnitudes. On the contrast, by nitrogen adsorption analysis commonly we could not measure macropores, larger than 300 μm. Therefore, the results from nitrogen adsorption and SEM could not be compared but rather discussed separately. However, together with gas adsorption analysis we could conclude, that in HM-pectin, LM-pectin and xanthan aerogels hydrophobic interactions cause denser cross-linking, resulting in increased micropore and mesopore volume. This phenomena is thus responsible for those materials being mechanically stable opposite from brittle alginate and guar aerogels.
 | ||
| Fig. 6 SEM images of (A) HM-pectin, (B) LM-pectin, (C) xanthan gum, (D) alginate and (E) guar gum. | ||
3.2. Thermal properties of aerogels
Measuring the thermal conductivity the results coincide with the surface area and porosity measurements. Namely, as the surface area and porosity increased, the thermal conductivity decreased. Low (0.021 W m−1 K−1) thermal conductivity was observed for LM-pectin aerogel, as presented in Table 2. The conductivity is promising nevertheless it could be improved, for instance by making pectin–silica composites, similarly like that of cellulose–silica.31 Better conductivity of pectin aerogels was provided by Rudaz et al.7 (0.016–0.022 W m−1 K−1). Their results were in agreement with those, reported in this research, when comparing the conductivity as a function of a density. Another research24 reports ultralow conductivity of liquid-crystalline nanocellulose (0.018 W m−1 K−1). Recent research work shows also extraordinary properties of alginate aerogels,20 as their conductivity reached 0.018–0.022 W m−1 K−1. Otherwise the low thermal conductivity was observed only by inorganic silica aerogels. HM-pectin aerogels also provided low conductivity of 0.023 W m−1 K−1. The conductivity of other polysaccharides was higher, especially for alginate and guar, most probably due to the presence of macropores. The thermal conductivity of aerogels as a function of bulk density is presented in Fig. 7. The increase in thermal conductivity with the increase of the density is a known phenomenon. The actual conductivity in the solid does not change but the area of the solid in a cross-section of the material decreases. This results in lower solid conduction per square meter of the porous material. A decreased density will increase the heat flow due to radiation, which will counter the gain in the solid conduction. The gas conductivity in porous material can be decreased by decreasing the pore size of the material. The collisions between the gas molecules and the solid are elastic which transfer small amounts of energy compared to the collision between gas molecules. Smaller pores lead to a higher probability of collisions between pore walls instead of other gas molecules. This is called the Knudsen effect. Polysaccharide aerogels have been until now mainly used for pharmaceutical or biomedical applications. Those results on thermal conductivity show another possibility for using those materials, especially pectin, as super-insulators. | ||
| Fig. 7 Thermal conductivity as a function of bulk density. | ||
Thermal analysis was performed on TGA/DSC1 (Mettler Toledo) apparatus. It can be observed from Fig. 8 that HM-pectin, LM-pectin and alginate degrade almost at the same temperature, 230 °C. Xanthan gum then degrades a few degrees later at about 270 °C. The exothermic decomposition was observed by the HM-pectin, LM-pectin, xanthan and alginate aerogels. Guar gum behaves completely differently from the other aerogels since it melts at 300 °C.
 | ||
| Fig. 8 Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) of (a) HM-pectin (b) LM-pectin (c) xanthan gum (d) alginate and (e) guar gum aerogels. | ||
3.3. Mechanical properties
Rheology measurements were performed for wet gels. Alcogels were prepared as described in the experimental section, by forming the polysaccharide solution and then induce the gelation in ethanol. Gels were prepared in forms of disc/tablet. It is evident from Fig. 9 that the highest difference between the storage and the loss modulus was observed for LM-pectin gel. Here the loss modulus (G′′) was around 30% of the elastic modulus (G′) which indicates full elasticity of the system. Other polysaccharides gels provided similar results (G′ > G′′) although the difference between both was lower. When increasing the strains, the elastic structure breaks down. Consequently the elastic modulus decreases. At the point where the G′′ becomes greater than G′ it is a crossover strain, and here the elastic structure is broken. The loss modulus was similar for all the samples, the higher difference was observed in the elastic modulus. The elastic modulus decreased in order, starting with LM-pectin, HM-pectin, xanthan, and ending with alginate and guar. The same behavior was observed by the dry aerogels when measuring the surface area and porosity, as well as the thermal conductivity. Therefore it is clear that all those variables are connected and depend mostly on the type of polysaccharide. | ||
| Fig. 9 Strain sweep experiments of polysaccharide alcogels at constant frequencies. (A) HM-pectin, (B) LM-pectin, (C) xanthan, (D) alginate and (E) guar. | ||
4. Conclusions
Ethanol was used to induce the gelation of alginate, low-methoxyl, high-methoxyl pectins, xanthan and guar gum without additional cross-linkers. Conventionally, those polysaccharides are prepared by dissolution in water, cross-linking by different cross-linkers and then prior supercritical drying the water in gel has to be exchanged with organic solvent. Since in this research, the gelation occurred directly in ethanol without additional cross-linkers, there was also no need for the solvent exchange step. The total production time was thus reduced. Monoliths with large surface areas and high porosities were obtained. Highest yet reported surface area of monolithic aerogels was obtained for LM-pectin (510 m2 g−1), HM-pectin (384 m2 g−1), xanthan (363 m2 g−1) and guar (111 m2 g−1). The total production time was lower compared to conventional polysaccharide aerogel synthesis since in this process no solvent-exchange or washing was needed prior supercritical drying. In addition, to the best of our knowledge this is the first report on the preparations of pure xanthan and guar aerogels. Very low thermal conductivity was observed for LM and HM-pectin aerogels and thus those materials could also be considered as insulating materials.5. Acknowledgement
The authors want to acknowledge the Slovenian Research Agency (Grant number: 1000-11-860046) for its financial support. Special thanks goes to Mettler Toledo d.o.o., Slovenia for their help when performing the thermal conductivity measurements by the DSC. The authors are grateful to Anton Paar Ljubljana, Slovenia for performing the measurements on the MCR302 Rheometer.References
- G. Leone and R. Barbucci, in Hydrogels, Springer Milan, 2009, pp. 25–41 Search PubMed.
- C. A. García-González, M. Alnaief and I. Smirnova, Carbohydr. Polym., 2011, 86, 1425–1438 CrossRef PubMed.
- K. S. Mikkonen, K. Parikka, A. Ghafar and M. Tenkanen, Trends Food Sci. Technol., 2013, 34, 124–136 CrossRef CAS PubMed.
- A. Veronovski, G. Tkalec, Ž. Knez and Z. Novak, Carbohydr. Polym., 2014, 113, 272–278 CrossRef CAS PubMed.
- H.-B. Chen, B.-S. Chiou, Y.-Z. Wang and D. A. Schiraldi, ACS Appl. Mater. Interfaces, 2013, 5, 1715–1721 CAS.
- C. A. García-González, E. Carenza, M. Zeng, I. Smirnova and A. Roig, RSC Adv., 2012, 2, 9816–9823 RSC.
- C. Rudaz, R. Courson, L. Bonnet, S. Calas-Etienne, H. Sallée and T. Budtova, Biomacromolecules, 2014, 15, 2188–2195 CrossRef CAS PubMed.
- A. M. Stephen and G. O. Phillips, Food Polysaccharides and Their Applications, CRC Press, 2014 Search PubMed.
- R. J. White, V. L. Budarin and J. H. Clark, Chem.–Eur. J., 2010, 16, 1326–1335 CrossRef CAS PubMed.
- P. D. Gaudio, G. Auriemma, T. Mencherini, G. D. Porta, E. Reverchon and R. P. Aquino, J. Pharm. Sci., 2013, 102, 185–194 CrossRef PubMed.
- A. Veronovski, Ž. Knez and Z. Novak, J. Supercrit. Fluids, 2013, 79, 209–215 CrossRef CAS PubMed.
- A. Veronovski, Ž. Knez and Z. Novak, Int. J. Pharm., 2013, 454, 58–66 CrossRef CAS PubMed.
- H. H. Tønnesen and J. Karlsen, Drug Dev. Ind. Pharm., 2002, 28, 621–630 CrossRef PubMed.
- L. Wang, D. A. Schiraldi and M. Sánchez-Soto, Ind. Eng. Chem. Res., 2014, 53, 7680–7687 CrossRef CAS.
- D. Bilanovic, J. Starosvetsky and R. H. Armon, Food Hydrocolloids, 2015, 44, 129–135 CrossRef CAS PubMed.
- K. S. Mikkonen, K. Parikka, J.-P. Suuronen, A. Ghafar, R. Serimaa and M. Tenkanen, RSC Adv., 2014, 4, 11884–11892 RSC.
- T. Mehling, I. Smirnova, U. Guenther and R. H. H. Neubert, J. Non-Cryst. Solids, 2009, 355, 2472–2479 CrossRef CAS PubMed.
- R. Valentin, R. Horga, B. Bonelli, E. Garrone, F. Di Renzo and F. Quignard, Macromol. Symp., 2005, 230, 71–77 CrossRef CAS PubMed.
- R. Valentin, R. Horga, B. Bonelli, E. Garrone, F. Di Renzo and F. Quignard, Biomacromolecules, 2006, 7, 877–882 CrossRef CAS PubMed.
- P. Gurikov, S. P. Raman, D. Weinrich, M. Fricke and I. Smirnova, RSC Adv., 2015, 5, 7812–7818 RSC.
- F. Liebner, E. Haimer, M. Wendland, M.-A. Neouze, K. Schlufter, P. Miethe, T. Heinze, A. Potthast and T. Rosenau, Macromol. Biosci., 2010, 10, 349–352 CrossRef CAS PubMed.
- R. Gavillon and T. Budtova, Biomacromolecules, 2008, 9, 269–277 CrossRef CAS PubMed.
- R. Sescousse, R. Gavillon and T. Budtova, Carbohydr. Polym., 2011, 83, 1766–1774 CrossRef CAS PubMed.
- Y. Kobayashi, T. Saito and A. Isogai, Angew. Chem., Int. Ed., 2014, 53, 10394–10397 CrossRef CAS PubMed.
- M. Robitzer, A. Tourrette, R. Horga, R. Valentin, M. Boissière, J. M. Devoisselle, F. Di Renzo and F. Quignard, Carbohydr. Polym., 2011, 85, 44–53 CrossRef CAS PubMed.
- R. Baetens, B. P. Jelle and A. Gustavsen, Energ. Build., 2011, 43, 761–769 CrossRef PubMed.
- L. Ling and M. Qing-Han, J. Mater. Sci., 2005, 40, 4105–4107 CrossRef.
- S. J. Kim, S. W. Hwang and S. H. Hyun, J. Mater. Sci., 2005, 40, 725–731 CrossRef CAS.
- Z. Ulker and C. Erkey, J. Controlled Release, 2014, 177, 51–63 CrossRef CAS PubMed.
- I. Smirnova, in Aerogels Handbook, ed. M. A. Aegerter, N. Leventis and M. M. Koebel, Springer New York, New York, NY, 2011, pp. 695–717 Search PubMed.
- A. Demilecamps, C. Beauger, C. Hildenbrand, A. Rigacci and T. Budtova, Carbohydr. Polym., 2015, 122, 293–300 CrossRef CAS PubMed.
- D. Oakenfull and A. Scott, J. Food Sci., 1984, 49, 1093–1098 CrossRef CAS PubMed.
- A. N. Sabirzyanov, A. P. Il'in, A. R. Akhunov and F. M. Gumerov, High Temp., 2002, 40, 203–206 CrossRef.
- D. Bellet and L. Canham, Adv. Mater., 1998, 10, 487–490 CrossRef CAS.
- M. J. van Bommel and A. B. de Haan, J. Non-Cryst. Solids, 1995, 186, 78–82 CrossRef CAS.
- J. E. S. D. Ladbury, B. R. Currell, J. R. Horder, J. R. Parsonage and E. A. Vidgeon, Thermochim. Acta, 1990, 169, 39–45 CrossRef CAS.
- M. Hu, D. Yu and J. Wei, Polym. Test., 2007, 26, 333–337 CrossRef CAS PubMed.
- M. Merzlyakov and C. Schick, Thermochim. Acta, 2001, 377, 183–191 CrossRef CAS.
- S. M. Marcus and R. L. Blaine, Thermochim. Acta, 1994, 243, 231–239 CrossRef CAS.
- T. Lund, O. Smidsrød, B. Torger Stokke and A. Elgsaeter, Carbohydr. Polym., 1988, 8, 245–256 CrossRef CAS.
- M.-A. Lungan, M. Popa, J. Desbrieres, S. Racovita and S. Vasiliu, Carbohydr. Polym., 2014, 104, 213–222 CrossRef CAS PubMed.
- G. Copetti, M. Grassi, R. Lapasin and S. Pricl, Glycoconjugate J., 1997, 14, 951–961 CrossRef CAS.
- A. Veronovski, Z. Novak and Ž. Knez, J. Biomater. Sci., Polym. Ed., 2012, 23, 873–886 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |