
Electrokinetic remediation: challenging and optimization of electrolyte for sulfate removal in textile effluent-contaminated farming soil†
Abstract
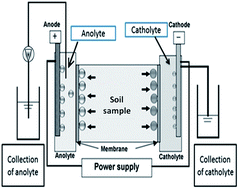
In the current situation, soil salinity has increased due to the uncontrolled discharge of industrial effluent. In the present investigation, an electrokinetic (EK) technique was used for the inclusion and removal of sulfate in farming soil. Initially, the influence of the electrode distance was evaluated by the use of two cell configurations. The nature of the catholyte was assessed for the inclusion of sulfate in the soil. Decolorized textile effluent and 5% Na2SO4 were tested as the catholyte and double-distilled water as the anolyte for the inclusion process. Subsequently, selection of the electrolyte concentration and pH was investigated for the removal of sulfate concentrations in contaminated farming soil. Optimization of the pH and electrolyte for the process of sulfate removal in farming soil was performed and a mechanism of the removal process was proposed. A total of 96% sulfate removal was achieved using 0.01 M hydrochloric acid at a pH of 4.5 for a 96 h EK experiment. The initial conductivity of the soil was 5.32 dS m−1, and after sulfate removal the conductivity was about 0.714 dS m−1 and the energy consumption was 119 kW h m−3 for the farming soil.
 Please wait while we load your content...
Please wait while we load your content...

