
Chirality transfer through sulfur or selenium to chiral propellers†
Abstract
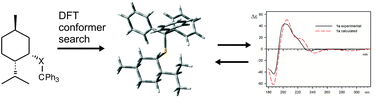
The mechanism of chirality transfer from a chiral alkyl substituent to a trityl moiety through sulfur or selenium atoms is analysed and discussed on the basis of ECD measurements, DFT structure and ECD spectra calculations. It is shown that the presence of a chalcogen atom is manifested by elongation of the distance between the chiral and the trityl moieties as well as by the change of electronic properties of the trityl chromophore while maintaining its chiroptical response to the chirality of the molecule.
 Please wait while we load your content...
Please wait while we load your content...

