
Design, synthesis and thermal behaviour of a series of well-defined clickable and triggerable sulfonate polymers†
Abstract
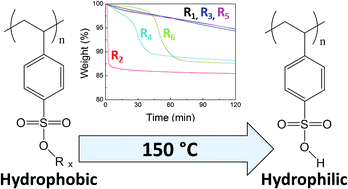
In the printing industry, the exploitation of triggerable materials that can have their surface properties altered on application of a post-deposition external stimulus has been crucial for the production of robust layers and patterns. To this end, herein, a series of clickable poly(R-alkyl p-styrene sulfonate) homopolymers, with systematically varied thermally-labile protecting groups, has been synthesised via reversible addition-fragmentation chain transfer (RAFT) polymerisation. The polymer range has been designed to offer varied post-deposition thermal treatment to switch them from hydrophobic to hydrophilic. Suitable RAFT conditions have been identified to produce well-defined homopolymers (Đ, Mw/Mn < 1.11 in all cases) at high monomer conversions (>80% for all but one monomer) with controllable molar mass. Poly(p-styrene sulfonate) with an isobutyl protecting group has been shown to be the most readily thermolysed polymer that remains stable at room temperature, and was thus investigated further by incorporation into a diblock copolymer, P3HT-b-PiBSS, by click chemistry. The strategy for preparation of thermal modifiable block copolymers exploiting R-protected p-styrene sulfonates and azide-alkyne click chemistry presented herein allows the design of new, roll-to-roll processable materials for potential application in the printing industry, particularly organic electronics.
 Please wait while we load your content...
Please wait while we load your content...

