
The non-innocent nature of graphene oxide as a theranostic platform for biomedical applications and its reactivity towards metal-based anticancer drugs†

Abstract
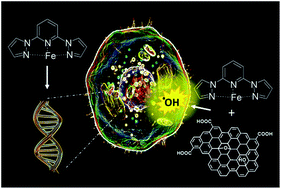
The self-assembly process in a solution of a mononuclear iron(II) complex based on the bispyrazolylpyridine scaffold with graphene oxide (GO) micrometer-sheets allows not only the devising of a new hybrid-architecture for GO-based materials suitable for nanomedicine, but also the unveiling of the reactive nature of GO as a drug-carrier. The neat iron complex is found to be highly active in disrupting the cell cycle through DNA binding, with behaviour and efficiency similar to that expressed by ruthenium-complexes as well as antibiotic-drugs such as doxorubicin. On the contrary, in the hybrid material the proclivity of neat GO to produce reactive oxygen species (ROS) became down-regulated by the electron-buffering properties of the loaded iron complex, evidencing the presence of an active electron transfer from the drug to GO. These findings question the use of the neat GO platform as a suitable carrier for metal-based anticancer drugs and highlight the importance of addressing the chemical/physical integrity of the drug being loaded into GO before drawing conclusions on the potential effectiveness of the hybrid material for medical applications.
 Please wait while we load your content...
Please wait while we load your content...

