
Effect of graphene on the self-assembly and rheological behavior of a triblock copolymer gel†
Mahla Zabet,
Satish Mishra and
Santanu Kundu*
Dave C. Swalm School of Chemical Engineering, Mississippi State University, MS, USA. E-mail: santanukundu@che.msstate.edu
First published on 28th September 2015
Abstract
The self-assembly behavior of physical gels and the effect of such an assembly on the mechanical properties are of significant interest. Here, graphene nanoplatelets have been incorporated in a triblock copolymer gel consisting of poly(methyl methacrylate)–poly(n-butyl acrylate)–poly(methyl methacrylate) [PMMA–PnBA–PMMA] in a midblock selective solvent, 2-ethyl-1-hexanol. The thermoreversible nature of the gel allowed us to incorporate the graphene nanoplatelets in a liquid stage and these nanoplatelets then become part of the network structure as the solution is cooled. Shear rheology is used to investigate the change of mechanical properties as a function of graphene concentration. Graphene nanoplatelets affect the self-assembly behavior as demonstrated by the decrease of gelation temperature. Interestingly, no significant increase of modulus was observed with the incorporation of graphene, as typically observed in polymer nanocomposites. This indicates that the graphene nanoplatelets are not actively participating in load-bearing, i.e. the platelets are not elastically active. A small change of relaxation time in graphene containing triblock copolymer gels has been observed, as captured by stress relaxation experiments. Our results provide a method to incorporate nanomaterials in physically crosslinked polymer gels without affecting the mechanical properties significantly.
Introduction
The addition of nanomaterials in a polymeric matrix can result in the improvement of mechanical properties, thermal and electrical conductivities.1 It is often possible to render a nanocomposite multifunctional by selecting nanoparticles of suitable functionalities and by controlling the hierarchical structure from the nano to micro scale.2,3 A similar level of (multi) functionalities can be achieved in gels and hydrogels but such attempts have been limited. Selecting nanoparticles with desired functionalities and then incorporating these nanoparticles in polymer gels can potentially result in materials with novel and interesting properties.4–7A typical polymer gel consists of two phases, polymer and solvent. The nanoparticles can have different affinities to these phases and can preferentially interact with one of these phases. Such interactions can result in a different gel structure – particularly for the physical gels – compared to that of the pristine gel without nanoparticles. Similar to polymer nanocomposites, changes in gel structure expect to result in change of properties. Here, we report the effect of addition of few layers graphene on the self-assembly behavior and mechanical properties of a physically associating gel.
In a gel or swollen polymer network, polymer strands are connected at the junctions (cross-links) and the elastic modulus of the gel depends on the number of load-bearing strands per unit volume.8 If nanomaterials are incorporated in a gel, the added nanomaterials can be a part of the network in different ways. For example, these nanomaterials can act as crosslinkers or can themselves form a percolation network.4,9 For example, a poly(n-isopropylacrylamide) (PNIPA) hydrogel with nanoclay as a crosslinker displays improved mechanical strength and extensibility compared to the gels without nanoclay and prepared by free-radical polymerization.10,11 The improved properties of these gels was attributed to the way the polymer chains are connected to the clay particles.10,11
Similar trend has also been observed in graphene containing polyacrylamide gels. Das et al. have investigated the effect of pristine graphene on mechanical properties of polyacrylamide hydrogels. The addition of graphene resulted in increase of modulus in comparison to a gel without graphene. In this study, high molecular weight polyacrylamide was used as a stabilizer for graphene preparation. These long polymer chains can wrap around the graphene nanoplatelets or physisorbed on the graphene surfaces.4 During the gelation process, the polymer chains present in water can entangle with the polymer chains associated with the graphene nanoplatelets. As a result, the graphene nanoplatelets become part of the network. During mechanical deformation process both polymer chains and graphene platelets participate in load bearing. This phenomena results in increase of modulus. At higher strain the polymer chains gradually disentangle, as manifested by step responses of stress–strain curves.
Gelation process of two-dimensional nanoparticles in a solvent can take place without any polymer phase, if a percolation threshold is reached. For example, aqueous solution of graphene oxide nanoplatelets for a concentration of as low as 0.05 wt% can form gels.9,12
Recent literature shows importance of graphene because of their remarkable electronic, thermal, and mechanical properties.13 The unique properties of graphene motivates us to incorporate it in a physically associating thermoreversible gel, in which a viscous polymer solution forms a gel with the decrease of temperature.14–18 The thermoreversible nature of the gel allows us to integrate the graphene nanoplatelets in the liquid phase and as the sample is cooled, the nanoplatelets be part of the gel network. It is anticipated that the addition of graphene will make these gels electroactive.
Efficient exfoliation of graphene from graphite flakes is a significant challenge because of π–π stacking of the graphene layers.19 Liquid phase exfoliation,19 micromechanical cleavage,20 chemical vapor deposition,21 and epitaxial growth on SiC substrates22 are the most common methods for graphene production. Although all these techniques provide several advantages, the direct liquid phase exfoliation of graphite was used in this study. This technique yielded high concentration of graphene nanoplatelets in the solvent.
The physical gel considered here consists of a triblock copolymer, poly(methyl methacrylate)–poly(n-butyl acrylate)–poly(methyl methacrylate) [PMMA–PnBA–PMMA], in a midblock selective solvent, 2-ethyl-1-hexanol. This is a well-studied system,14–16 where, at elevated temperature the polymer become soluble in 2-ethyl-1-hexanol. However, as the temperature is decreased, the solubility of PMMA blocks in the solvent decreases. In fact, the solvent interaction parameter, χ, for PMMA in alcohol (such as 2-ethyl-1-hexanol) is highly temperature dependent.14,23 At low temperature, a number of collapsed endblocks of PMMA self-assemble to form aggregates. These aggregates are connected by PnBA chains and a three-dimensional gel is obtained. Here, we report the effect of graphene nanoplatelets on the physical gelation process. Two different concentrations of graphene nanoplatelets suspended in 2-ethyl-1-hexanol have been considered. Addition of graphene platelets decreases the gelation temperature, however the mechanical properties of the graphene containing gels are similar to that of the pristine gels at a temperature far below the gelation temperature.
Experimental
The graphene dispersions were prepared by exfoliating expanded graphite, kindly provided by Asbury Carbons (CAS # 7782-42-5, grade 3806), in 2-ethyl-1-hexanol (Fisher Scientific). Expanded graphite was added to 2-ethyl-1-hexanol at two different concentrations: 0.5 mg mL−1 and 1 mg mL−1. A tip sonicator (Fisher Scientific, CL-334) at 80 W for 10 h was utilized to exfoliate the graphite. An ice-bath was used to avoid the temperature increase of the solution due to prolonged sonication. The dispersion was centrifuged (Eppendorf, 5810 R) at 4000 rpm for 30 min. The centrifuge process resulted in the larger, non-exfoliated graphite particles to separate out of the solvent and the exfoliated nanoplatelets containing few-layers of graphene remained in the solution phase. The supernatant has been decanted for further use and characterization. To determine the graphene concentration in the supernatant, filtration technique was used. Here, the supernatant was pushed through a 0.2 μm PTFE filter (Millipore) to filter out the graphene nanoplatelets. The filter was then dried overnight in vacuum and the increase of mass of the filter was measured. The 0.5 mg mL−1 and 1 mg mL−1 concentration of graphite resulted in 0.04 mg mL−1 and 0.12 mg mL−1 graphene in the supernatant. To analyze the stability of graphene dispersions, a spectrophotometer (Unico, model # 1100) was used. The absorbance at a wavelength of 660 nm was measured.Gels were prepared by dissolving a triblock copolymer PMMA–PnBA–PMMA (kindly provided by Kuraray Co) in graphene dispersion at a temperature of 80 °C. The triblock copolymer consists of two poly(methyl methacrylate) end blocks with molecular weight of 9000 g mol−1 which are separated by a poly(n-butyl acrylate) midblock having a molecular weight of 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Based on the product datasheet, the triblock copolymer has a polydispersity in the range of 1.2 to 1.4. Gel formation took place when the polymer solution was cooled to room temperature. For the present study the polymer volume fraction was considered to be 5 vol% (0.0539 mass fraction).
000 g mol−1. Based on the product datasheet, the triblock copolymer has a polydispersity in the range of 1.2 to 1.4. Gel formation took place when the polymer solution was cooled to room temperature. For the present study the polymer volume fraction was considered to be 5 vol% (0.0539 mass fraction).
Both standard and cryo-TEM were used to characterize the graphene samples and graphene containing gel samples. TEM measurements were conducted using a 200 kV JEOL 2100 instrument on samples prepared by drop casting of a few drops of dispersion on to the carbon-film-covered copper grids. Cryo-TEM image was obtained using 200 kV Mark IV by FEI Co (at Tulane University). A drop of 4 microliter liquid sample was placed on a 200 mesh lacey carbon grid. The grid was lifted into an 100% humidity chamber and blotted by filter paper from both sides and then plunged into liquid ethane (−140 °C) to freeze and to vitrify the samples. The grid was transferred in liquid nitrogen to a cryo-TEM holder through cryo-transfer station. The sample was investigated at −170 °C.
To measure the thickness of the graphene nanoplatelets, atomic force microscopy (AFM) was conducted on the graphene nanoplatelets deposited on mica substrate. A drop of graphene containing 2-ethyl-1-hexanol was placed on a mica substrate. The sample was dried in vacuum. Imaging was conducted using Dimension Icon AFM (Bruker Corporation). Silicon nitride probe with the spring constant of 0.4 N m−1 was used.
The rheological characterization of graphene gels was performed using a Discovery HR-2 hybrid rheometer with 25 mm diameter parallel plate geometry. A gap of 1 mm was maintained. The temperature-dependent dynamic moduli of these gels were measured at strain amplitude of 5% by varying the temperature from 55 °C to 0 °C at a rate 2 °C min−1. The strain-sweep experiments at 6 °C and 22 °C were conducted by varying strain amplitude from 0.1% to 300% at a frequency of 1 rad s−1. The steady-shear viscosities were measured at 50 °C using a cone and plate geometry. All experiments were repeated at least three times.
Results and discussion
A stable solution of exfoliated graphene nanoplatelets in 2-ethyl-1-hexanol is necessary to achieve the uniform incorporation of graphene nanoplatelets in the triblock copolymer gels. Fig. 1a displays 2-ethyl-1-hexanol with 0.04 mg mL−1 and 0.12 mg mL−1 of graphene, corresponding to graphene mass fractions of 0.44 × 10−4 and 1.3 × 10−4, respectively. These solutions are the supernatant of the graphene containing centrifuged samples. Pure solvent is also shown for comparison purpose. To investigate the stability of the suspensions, the samples were stored for 7 days without any perturbation. As shown in Fig. 1b, the solution color remained unchanged after 7 days of waiting, indicating the stable nature of the solution. To investigate further, visible light absorbance spectrophotometry was used.4,24 Here, absorbance by the solution at a wavelength of 660 nm was measured on day #1 and on day #7. In both cases the samples were diluted by a factor of 5 before measuring the absorbance to avoid the saturation of the spectrophotometer detector. As shown in Fig. 1c, a small decrease in absorbance of graphene dispersions (6.23% for 0.04 mg mL−1 and 6.7% for 0.12 mg mL−1) was observed further confirming the stability of these solutions after even seven days. | ||
| Fig. 1 (a) Solvent without and with few layers of graphene (FLG) on (a) day #1 and (b) day #7. (c) Absorbance at λ = 660 nm for the samples with 0.04 mg mL−1 and 0.12 mg mL−1 FLG on day #1 and day #7. In both cases, the samples were diluted by a factor of 5. The error bars represent one standard deviation. | ||
In general, good exfoliation of graphite in a solvent is achieved when the surface energy of the solvent is similar to that of graphene. It has been shown that the solvents with the surface energy in the range of 40–50 mJ m−2 are good solvents for graphite exfoliation.25 The Hildebrand solubility parameters can also be used to choose an appropriate solvent for dispersion. The dispersive Hansen solubility parameter is only considered due to the nonpolar nature of graphene.26 It has been shown that solvents with the dispersive solubility parameter in the range of 15–21 MPa1/2 are good solvents for graphene.26 The surface tension of 2-ethyl-1-hexanol is close to 27 mJ m−2, which is not close to the graphite surface energy. However, the dispersive solubility factor of 2-ethyl-1-hexanol is 16 MPa1/2, which makes it a good solvent for graphene exfoliation.
An achieved graphene concentration of 0.12 mg mL−1 in this study is comparable to that obtained for other solvents reported in the literature.27,28 For example, graphene concentration of as high as 1.2 mg mL−1 in NMP and 0.5 mg mL−1 in isopropanol has been obtained after prolonged sonication (more than 100 hours). However, prolonged sonication has shown to cause damage of graphene structure due to high pressure and temperature produced by cavitation.27,28 In an alternative approach, to obtain higher graphene concentration, the graphene platelets have been stabilized using polyvinylpyrrolidone (PVP).29
Stability of a graphene suspension depends on the settling velocity of the graphene platelets in the static suspending fluid. The separation process that takes place during the centrifugation also depends on the settling velocity.30 Based on Stokes' law, the settling velocity, uT, for spherical particles can be expressed as, uT = d2(ρP − ρf)g/18μ.31 Here, d is the diameter of a particle, ρP and ρf are the densities of the particle and the suspending fluid, respectively, g is the acceleration due to the gravity and μ is the viscosity of suspending fluid.31 The equation for nanoplatelets will have a different prefactor than that obtained for spherical particles. However, the key factors for terminal velocity are the difference in the density of the particles and the fluid, and the fluid viscosity. Although, the surface tension of 2-ethyl-1-hexanol is similar to ethanol and isopropanol, the density and viscosity of 2-ethyl-1-hexanol are higher than of those. This likely resulted in higher concentration of graphene nanoplatelets in 2-ethyl-1-hexanol for a relatively short sonication time. The graphene dispersed solution behaves like a liquid, and gelation due to percolation of graphene nanoplatelets has not been observed.
Once a stable solution is obtained, polymer pellets were added in the solutions to obtain polymer volume percentage of 5%. Fig. 2a displays the gels without and with graphene. At room temperature both the solutions, without and with graphene, form gel, as tested by vial-inversion tests (Fig. 2a). Dark color represents the presence of graphene in the system. No visible heterogeneities were observed in graphene containing gels. Also, similar to the pristine gels (without graphene), multiple heating and cooling cycles did not lead to any change of rheological properties of the graphene containing gels.
 | ||
| Fig. 2 (a) Gels with and without graphene. Polymer concentration is maintained at 5 vol%. (b) Cryo-TEM image of a gel containing 0.12 mg mL−1 FLG. (c) Temperature dependence of storage (G′) and loss moduli (G′′) as a function of temperature for gels with and without FLG. The error bars represent one standard deviation. | ||
To investigate the level of exfoliation of graphite, TEM and cryo-TEM techniques were used for graphene containing solvents and gels. Fig. 2b displays a typical graphene nanoplatelet present within a gel obtained using cryo-TEM. The application of cryo-TEM is advantageous, as evaporation of 2-ethyl-1-hexanol does not take place during the sample preparation stage and the gel structure is preserved. Although polymer phase cannot be resolved, the graphene nanoplatelets are distinctly visible. The graphene platelets are found to be folded with lateral dimension higher than 500 nm. Similar observation has been made for the graphene nanoplatelets dispersed in 2-ethyl-1-hexanol (Fig. S1 and S2 in ESI†). We also attempted to determine the thickness of nanoplatelets, i.e., the number of graphene layers present in one platelet. Images of the edge of the platelets were collected (Fig. S3 in ESI†) and the number of layers was counted for at least 20 samples for each graphene concentration. The distributions of number of graphene layers present in the nanoplatelets are shown in Fig. S1 and S2 in ESI.† Electron diffraction patterns were also analyzed at different spots on graphene platelets samples. Single layer and folded single layer mostly display first order peak, whereas, second or multiple order reflections were observed for multiple layers.32 Simulation study indicates that for single layer of graphene, the intensity of inner spots is higher than that observed for outer spots.25,32 Conversely, for multilayers of graphene, the intensity of outer spots is more than that is observed for inner spots.25,32 This can be quantified by comparing the intensity of the {1100} and {2110} diffraction peaks and the intensity ratio, I1100/I2110, greater than 1 indicates the presence of single layer graphene (Fig. S1 in ESI†).
To analyze the thickness of graphene layers, AFM experiments were conducted on deposited graphene nanoplatelets on mica surface from the solutions of different graphene concentration. Fig. 3 shows the AFM results for graphene nanoplatelets deposited from the solution containing 0.12 mg mL−1 of graphene. Similar to TEM observation, the graphene nanoplatelets have lateral dimensions in the order of 500 nm and some of the graphene layers are founded to be folded. The height profiles were obtained at multiple locations and the results indicate that the thickness of these layers is in the range of 2.5 nm to 4.3 nm. This corresponds to presence of approximately 7–12 graphene layers.
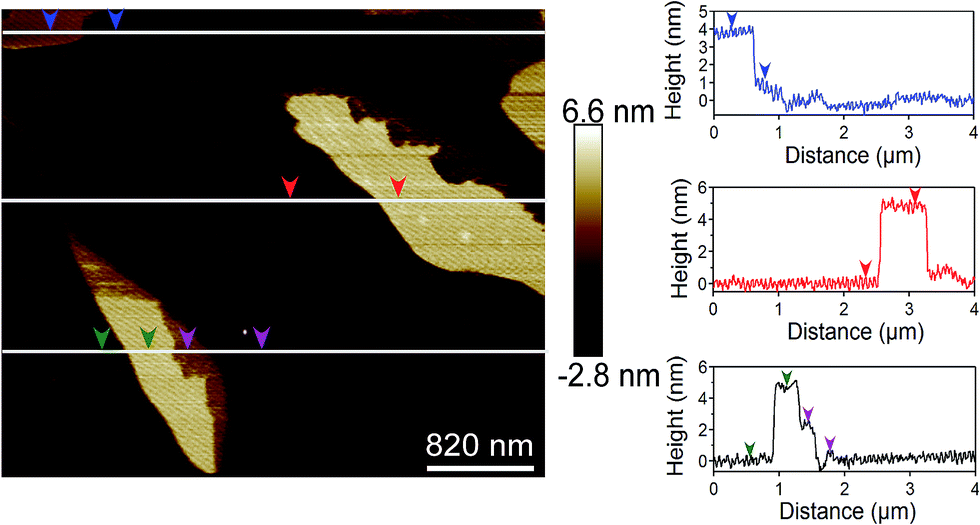 | ||
| Fig. 3 AFM image of graphene nanoplatelets on mica surface deposited from a suspension containing 0.12 mg mL−1 graphene. The height profiles were measured at multiple locations. | ||
Based on the TEM results, electron diffraction patterns and AFM data, it was found that the graphene nanoplatelets in our samples have varying thickness, from a single layer to as high as 12 layers. Interestingly, lower initial graphite concentration yielded more single layer graphene. For the solution containing 0.04 mg mL−1 graphene, the suspended graphene nanoplatelets have 1–3 layers, with approximately 30% single layer. For the graphene concentration of 0.12 mg mL−1, the graphene nanoplatelets have 6–12 layers. We have denoted our nanoplatelets as few-layer graphene (FLG), because of the presence of multiple graphene layers (as high as 12).
The effect of graphene on the gelation behavior of graphene containing polymer solution was quantified using oscillatory shear rheology. The samples were loaded in the rheometer at 55 °C in liquid form. Subsequently, the temperature was decreased at a rate of 2 °C min−1. During this process the sample was subjected to a strain of 5% and the storage and loss moduli were obtained from the rheometer software. The storage modulus (G′) represents the elastic contribution, whereas, the loss modulus (G′′) represents the viscous contribution. The results for a sample without graphene and two samples containing 0.04 mg mL−1 and 0.12 mg mL−1 FLG are shown in Fig. 2c. Although, at high temperature (above 40 °C), G′ is higher than G′′, unfortunately, the measured torque values at these temperatures are low and close to the instrument limit. This restricts us to evaluate the elasticity of the solutions at that temperature. However, the solutions visually appeared to be viscous liquids. Flow sweep experiments at 50 °C indicate that the viscosity of these solutions are low and the addition of graphene did not result into a significant change of viscosity values (Fig. S4 in ESI†).
As the temperature is decreased, the torque values reached the measurement limit and it was found that G′′ is higher than G′, i.e., the solution is a viscous liquid. With further decrease of temperature, both G′ and G′′ increase and a cross-over between G′ and G′′ was observed. Below the cross-over temperature, G′ remains higher than G′′, indicating that the viscous liquid self-assemble into a soft-solid or gel like material. The crossover temperature is defined as the gel point for our samples.14,16 For a sample without graphene, the gelation temperature is ≈32 °C. However, addition of FLG resulted in lower gelation temperature. For a sample containing 0.04 mg mL−1 graphene, the gelation temperature is ≈26 °C, whereas, for 0.12 mg mL−1 the gelation temperature further reduced to ≈23 °C. The decrease in gel point with incorporation of graphene confirms the effect of graphene on the self-assembly process. The graphene nanoplatelets hinders the network formation, which results in decrease of gelation temperature.
The triblock gel considered here displays unique strain-stiffening behavior, i.e., for an applied frequency the storage modulus increases with increasing strain amplitude.15,18 The strain-stiffening behavior is related to the finite-chain extensibility of PnBA chains. We also investigated the effect of FLG on the strain-stiffening behavior. Fig. 4a and b display the G′ and G′′ as a function of strain amplitude for pristine and graphene containing gels at 22 °C and 6 °C, respectively. At both of these temperatures strain-stiffening behavior is observed.
 | ||
| Fig. 4 Storage (G′) and loss moduli (G′′) as a function of strain amplitude for gels with and without FLG at (a) 22 °C and (b) 6 °C. The applied frequency was 1 rad s−1. | ||
Similar to that observed in temperature sweep experiments presented in Fig. 2, the G′ and G′′ values are different for different gel samples at 22 °C (Fig. 4a). For the pristine gel, the G′ is approximately 1 order of magnitude higher than G′′, indicating the elastic nature of this gel. However, with addition of FLG the difference between G′ and G′′ decreases. The decrease in the difference between storage and loss moduli for graphene containing gels confirms the effect of graphene on the self-assembly process.
Fig. 4b shows G′ and G′′ for these gels at 6 °C. Here, G′ and the difference between G′ and G′′ are relatively independent of the graphene concentration. In fact, as observed in Fig. 2c, the storage moduli for both pure and graphene containing gels are similar below 10 °C. It is likely that at that temperature the self-assembly process is adequate for the modulus to reach the values observed for pristine gels. It is most likely that the graphene platelets do not participate in load-bearing, i.e., those are not elastically active. This observation is different than that generally observed in literature, where addition of graphene resulted in increase of elastic modulus.4
Interestingly, at both 22 °C and 6 °C, the addition of FLG does not have any effect on the onset of the nonlinear elastic behavior of these gels and the strain stiffening response become apparent at an approximate strain value of 100% for both these temperatures.
To understand the self-assembly process further, we studied the relaxation behavior of these gel samples at different temperatures ranging from 6 °C to 25 °C. Here, a step strain of 5%, which falls within the linear viscoelastic region, was applied and the stress relaxation was captured as a function of time. From these experiments, time dependent modulus values G(t) are estimated as a function of time and are shown in Fig. 5. Overall, G(t) decreases with time indicating that the sample relaxes with time. The rate of change of G(t) depends on the experimental temperature and the sample tested. As expected, at lower temperature, where the sample is more elastic, the relaxation process is slower than that observed at higher temperature. Near the gelation point the relaxation is very rapid. Stress relaxation in the physical gels considered here takes place by the exchange of the endblocks in and out of the aggregates and between the neighbouring aggregates. Therefore, both temperature and graphene platelets affect the relaxation process.
 | ||
| Fig. 5 Stress relaxation of gels without graphene (a) and gels with 0.12 mg mL−1 graphene (b) studied over a temperature range of 6 to 25 °C. The symbols are experimental data, whereas the lines are model fit (eqn (1)). | ||
The stress-relaxation results can be fitted with different models, such as Maxwell model, stretched exponential function, Maxwell–Jeffrys, and Kelvin–Voigt models.8 Fitting with these models provide us information on relaxation time and its distribution, which are related to viscoelasticity and the self-assembly process. Here, the stress-relaxation data are fitted with a stretched exponential function given as:14,33
 | (1) |
Table 1 displays the fitted values for G0, τ, and β for the samples without and with graphene at different temperatures. The modulus values increases with decreasing temperature. This behavior is expected because of the less swollen state of the aggregates at lower temperature. Also, at lower temperatures, chain pull-out from the less swollen aggregates is difficult than more swollen states at higher temperatures. Therefore, the relaxation process becomes faster with increase of temperature and the change of relaxation time was almost two orders of magnitude.
| (a) | (b) | ||||||
|---|---|---|---|---|---|---|---|
| T | G0 (Pa) | τ (s) | β | T | G0 (Pa) | τ (s) | β |
| 6 °C | 202 ± 11 | 223 ± 33 | 0.23 | 6 °C | 218 ± 7 | 198 ± 3 | 0.27 |
| 10 °C | 196 ± 8 | 120 ± 34 | 0.22 | 10 °C | 186 ± 20 | 60 ± 14 | 0.29 |
| 15 °C | 175 ± 12 | 21 ± 6 | 0.23 | 15 °C | 161 ± 10 | 15 ± 6 | 0.27 |
| 20 °C | 135 ± 12 | 7 ± 2 | 0.25 | 20 °C | 119 ± 15 | 3 ± 2 | 0.27 |
| 25 °C | 78 ± 15 | 4 ± 2 | 0.3 | ||||
In previous studies, the stress relaxation behavior of a similar gel considered here (without graphene) in both linear and nonlinear regime have been reported.14,33 Erk and Douglas did not consider G0 as a fitting parameter, but used the storage modulus value (G′) at the frequency of ω = 100 rad s−1. In the present study, we conducted the experiments using a TA Instruments HR-2 hybrid rheometer and the maximum frequency that we can achieve in this instrument was 30 rad s−1.15 Beyond this frequency, the inertial effect becomes significant (the raw phase angle values are more than 150°). Thus, the three parameters fitting were used for our experimental data. The data was fitted with stretched exponential function from t = 1 s. The modulus values obtained for the pristine gel were found to be similar to that obtained by Erk and Douglas.
Erk and Douglas have reported β = 0.33 for the pristine gels. However, in our study the stretching exponent has found to be in the range of 0.2–0.3 for both the gels with and without graphene. Our results are similar to β = 0.2 obtained by Hotta et al. for a triblock gel consists of polystyrene–polyisoprene–polystyrene.34 Interestingly, Drzal and Shull obtained β = 0.53.14 The differences are probably due to the different instruments and experimental protocols utilized in these works.
Stress relaxation results indicate that both pristine and graphene containing gels display similar stress-relaxation behavior. However, at lower experimental temperature, with increasing graphene concentration, for example, at 15 °C and 20 °C the values of G0 and τ values are lower for graphene containing gels. At 25 °C, which is just above the gelation temperature for the graphene containing gel, the relaxation behavior is different compared to the pristine gel. This is likely due to liquid like behavior at this temperature. Stretched-exponential function cannot be fitted with the experimental data at 25 °C. For graphene gel, the decrease of the relaxation time with increasing temperature indicates the easier exchange of PMMA blocks in and out of the aggregates.
To investigate whether the fitted stretched exponential function has captured the experimental data adequately, creep experiments were conducted on these samples. Note that a stress-controlled rheometer is used in this study, which is well suited for creep experiments. For the creep experiments a stress of 100 Pa was applied and the creep compliance values are reported.
The creep compliance of the gel without and with 0.12 mg mL−1 graphene at 6 °C is presented in Fig. S5 in ESI.† Distinct creep-ringing was observed at short time scale, which gradually faded out. The long term creep compliance data (t > 5 s) was fitted with the stretched exponential function. We only considered the long term creep compliance, as the functional form for the stretched exponential function used here cannot capture creep ringing. A reasonable fit (Fig. S6 in ESI†) was obtained and the fitted values for τ, and β are similar to that obtained from the fitting of stress-relaxation data (Table 1). However, G0 was lower than that obtained in stress-relaxation experiments, indicating that the gel behaves softer in the stress controlled mode experiments. Interestingly, the creep ringing of the gels can be reasonably captured using Maxwell–Jeffreys model composed of springs, dashpots and inertia terms (Fig. S7 in ESI†).35 However, the model did not capture the long term behavior well, as the Maxwell–Jefferys model predicts much faster creep.
The above results indicate that the graphene nanoplatelets affect the self-assembly process. Next, we try to hypothesize the location of graphene nanoplatelets in the gel. In a previous study, Schoch et al. have investigated the effect of single-walled carbon nanotubes (SWNTs) on mechanical properties of triblock copolymer gel, similar to the one studied here.5 The nanotube mass fraction was varied from 1.2 × 10−4 to 5.8 × 10−3, higher than the graphene mass fractions of 0.44 × 10−4 and 1.3 × 10−4 considered here. It was assumed that the PMMA chains wrap around the SWNTs, which is very likely owing to their diameter in the order of 1 nm. Interestingly, although a higher mass fraction was considered, the addition of SWNT did not cause any difference in gelation temperature and mechanical properties. The elastic modulus of SWNT containing gels were similar to that was obtained for the pristine gel. Therefore, SWNTs did not affect the self-assembly process significantly.
The difference is self-assembly process between pristine and graphene containing gels can be intuitively linked to the diffusivities of the nanotubes and graphene. However, the diffusivity of nanotubes is higher than that of flat, graphene nanoplatelets. The relative difference in diffusivities can be estimated considering dilute solutions of these. The ratio of diffusivity of a plate-like particle to that of rod can be given by D0,plate/D0,rod = πL3/(4D3[ln(L/d) − 0.8]).36 Here, D0,plate and D0,rod are the diffusivities of a plate and a rod in a dilute solution, respectively. D is the equivalent diameter of a plate, L is the length of a rod and d is the diameter of a rod.40 If we consider D ∼ 500 nm (as observed in TEM micrographs), d ∼ 1 nm, and L ∼ 100 nm, the D0,plate/D0,rod = 0.00165, i.e., the diffusivity of a nanotube will be three orders of magnitude higher than that of a graphene platelet. Therefore, the difference in self-assembly process in graphene containing gels is likely affected by the size and shape of the nanoplatelets, and polymer–graphene interaction, but not by the diffusivity of the nanoplatelets.
Gel formation can be discussed using the solubility parameters of the constituting components.37 The solubility parameters of the solvent, homopolymer components of the triblock copolymer, and graphene nanoplatelets are shown in Table 2.26,38,39
| Name | δt | δd | δp | δh |
|---|---|---|---|---|
| Poly(methyl methacrylate) | 22.69 | 18.64 | 10.52 | 7.51 |
| Poly(n-butyl acrylate) | 19.55 | 16.38 | 8.97 | 5.77 |
| 2-Ethyl-1-hexanol | 20.1 | 16 | 3.3 | 11.9 |
| Graphene | 21.67 | 18 | 9.3 | 7.7 |
The solubility parameters of graphene nanoplatelets are similar to that of PMMA and 2-ethyl-1-hexanol. This indicates that the graphene nanoplatelets will have favorable interaction with PMMA and graphene nanoplatelets can act as favorable adsorption sites for PMMA endblocks. The affinity of PMMA chains to the graphene surfaces have been reported in a number of studies.40,41 It has been shown that due to favorable interaction, the chain mobility near the graphene surface decreases. The mobility is further restricted, if the graphene is functionalized. Such decrease of mobility resulted in higher glass transition temperature in some cases.40,41 In our sample, although we observe affinity of PMMA chains to the graphene platelets, restricted mobility of the chains was not obvious, which would have been manifested by increase of gelation temperature. This is probably because of the solvated state of the PMMA aggregates.
Fig. 6a displays the proposed structure of the pristine gel. As discussed above, solubility of PMMA endblocks in 2-ethyl-1-hexanol decreases drastically with decreasing temperature. The PMMA endblocks collapse and a number collapsed endblocks self-assemble to form aggregates (Fig. 6a). The PMMA aggregates are connected by the PnBA bridges forming a gel.16 It is important to note that the aggregates are not permanent in nature and an exchange of polymer chains from solution to the aggregates and vice versa takes place dynamically. The rate of exchange decreases with decreasing temperature, which has manifested by increasing relaxation time as the temperature is reduced from the gelation temperature. It has been hypothesized that the aggregates become glassy (i.e., the aggregates become frozen) at low enough temperature.33 At that temperature, both the modulus and relaxation time will not be a strong function of temperature, as noted in Table 1.
 | ||
| Fig. 6 Proposed structure of the triblock copolymer gel (a) without and (b) with graphene. | ||
The graphene platelets affect the gel structure and the proposed gel structure is shown in Fig. 6b. Due to short chain length (in the order of 20 nm), the PMMA endblocks cannot wrap around the graphene nanoplatelets having dimensions in the order of 500 nm, except at the edges (Fig. 6b). Therefore, the PMMA aggregates that formed with decreasing temperature physisorbed at the graphene surface. These aggregates are connected by the PnBA midblocks and a three-dimensional gel network is formed. The interaction between graphene and PMMA chains is not expected to be strong because of solvated state of the PMMA aggregates. With decreasing temperature the solvent is expelled from the aggregates and the interaction become stronger.
In a polymer network where the chains are not entangled, the stress is transferred by elastically active chains connecting the aggregates. In a pure gel transfer of stress occurs unhindered. However, the addition of graphene caused hindered stress transfer. Also, the strength of interaction between the PMMA aggregates and the graphene become important. As a result, the gelation for graphene gels occurs at a lower temperature. However, at lower temperature (<10 °C), when the interaction between graphene and PMMA become stronger and the aggregates become glassy, the modulus of graphene containing gels become similar to that of pristine gel.
In this paper we have shown that graphene nanoplatelets can be incorporated in a physical gel. At lower temperature, far away from the gelation temperature, the modulus of graphene containing gels is similar to that observed for pristine gels. This signifies that the nanoparticles can be incorporated in physical gels without changing the mechanical properties. This will potentially lead to multifunctional gels having similar mechanical properties as the pristine gels. The future research will involve the investigation of the responsiveness of these gels subjected to electric field.
Concluding remarks
Thermoreversible physical gels with graphene nanoplatelets have been prepared and a gel structure has been proposed based on the experimental results. The gelation temperature decreases with increasing graphene concentration, however, far below the gelation temperature the elastic modulus is independent of graphene concentration. The stress relaxation response of the gels was described using stretched exponential function in the temperatures range of 6 °C to 25 °C. The long term creep responses of the gels were also fitted using the stretched exponential function and it was found that the gel is softer in the stress-controlled mode experiments. Responsiveness of these gels subjected to electric field will be investigated in a future research.Acknowledgements
We wish to acknowledge financial support from the National Science Foundation [DMR-1352572] and [IIA-1430364].References
- D. R. Paul and L. M. Robeson, Polymer, 2008, 49, 3187–3204 CrossRef CAS PubMed.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef CAS PubMed.
- P. Akcora, H. Liu, S. K. Kumar, J. Moll, Y. Li, B. C. Benicewicz, L. S. Schadler, D. Acehan, A. Z. Panagiotopoulos, V. Pryamitsyn, V. Ganesan, J. Ilavsky, P. Thiyagarajan, R. H. Colby and J. F. Douglas, Nat. Mater., 2009, 8, 354–359 CrossRef CAS PubMed.
- S. Das, F. Irin, L. Ma, S. K. Bhattacharia, R. C. Hedden and M. J. Green, ACS Appl. Mater. Interfaces, 2013, 5, 8633–8640 CAS.
- A. B. Schoch, K. R. Shull and L. C. Brinson, Macromolecules, 2008, 41, 4340–4346 CrossRef CAS.
- J. Zhang, S. Xu and E. Kumacheva, J. Am. Chem. Soc., 2004, 126, 7908–7914 CrossRef CAS PubMed.
- C. Wang, N. T. Flynn and R. Langer, Adv. Mater., 2004, 16, 1074–1079 CrossRef CAS PubMed.
- M. T. Shaw and W. J. MacKnight, Introduction to Polymer Viscoelasticity, John Wiley & Sons, 2005 Search PubMed.
- O. C. Compton, Z. An, K. W. Putz, B. J. Hong, B. G. Hauser, L. Catherine Brinson and S. T. Nguyen, Carbon, 2012, 50, 3399–3406 CrossRef CAS PubMed.
- S. J. Banik, N. J. Fernandes, P. C. Thomas and S. R. Raghavan, Macromolecules, 2012, 45, 5712–5717 CrossRef CAS.
- K. Haraguchi, H.-J. Li, K. Matsuda, T. Takehisa and E. Elliott, Macromolecules, 2005, 38, 3482–3490 CrossRef CAS.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- P. L. Drzal and K. R. Shull, Macromolecules, 2003, 36, 2000–2008 CrossRef CAS.
- S. M. Hashemnejad and S. Kundu, Soft Matter, 2015, 11, 4315–4325 RSC.
- M. E. Seitz, W. R. Burghardt, K. T. Faber and K. R. Shull, Macromolecules, 2007, 40, 1218–1226 CrossRef CAS.
- N. Mischenko, K. Reynders, M. H. J. Koch, K. Mortensen, J. S. Pedersen, F. Fontaine, R. Graulus and H. Reynaers, Macromolecules, 1995, 28, 2054–2062 CrossRef CAS.
- K. A. Erk, K. J. Henderson and K. R. Shull, Biomacromolecules, 2010, 11, 1358–1363 CrossRef CAS PubMed.
- W. Du, X. Jiang and L. Zhu, J. Mater. Chem. A, 2013, 1, 10592–10606 CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- L. B. Biedermann, M. L. Bolen, M. A. Capano, D. Zemlyanov and R. G. Reifenberger, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 125411 CrossRef.
- M. Rubinstein and R. H. Colby, Polymer Physics, OUP, Oxford, 2003 Search PubMed.
- A. S. Wajid, S. Das, F. Irin, H. S. T. Ahmed, J. L. Shelburne, D. Parviz, R. J. Fullerton, A. F. Jankowski, R. C. Hedden and M. J. Green, ArXiv11071519 Cond-Mat, 2011.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- Y. Hernandez, M. Lotya, D. Rickard, S. D. Bergin and J. N. Coleman, Langmuir, 2010, 26, 3208–3213 CrossRef CAS PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. de and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- A. O'Neill, U. Khan, P. N. Nirmalraj, J. Boland and J. N. Coleman, J. Phys. Chem. C, 2011, 115, 5422–5428 Search PubMed.
- A. S. Wajid, S. Das, F. Irin, H. S. T. Ahmed, J. L. Shelburne, D. Parviz, R. J. Fullerton, A. F. Jankowski, R. C. Hedden and M. J. Green, Carbon, 2012, 50, 526–534 CrossRef CAS PubMed.
- L. A. Felton, Remington-Essentials of Pharmaceutics, Pharmaceutical Press, 2013 Search PubMed.
- R. B. Bird, W. E. Stewart and E. N. Lightfoot, Transport Phenomena, John Wiley & Sons, 2007 Search PubMed.
- Y. Zhang, L. Zhang, P. Kim, M. Ge, Z. Li and C. Zhou, Nano Lett., 2012, 12, 2810–2816 CrossRef CAS PubMed.
- K. A. Erk and J. F. Douglas, in Symposium LL/MM–Gels and Biomedical Materials, 2012, vol. 1418 Search PubMed.
- A. Hotta, S. M. Clarke and E. M. Terentjev, Macromolecules, 2002, 35, 271–277 CrossRef CAS.
- L. Pavlovsky, J. G. Younger and M. J. Solomon, Soft Matter, 2012, 9, 122–131 RSC.
- R. G. Larson, The Structure and Rheology of Complex Fluids, Oxford University Press, New York, 1st edn, 1998 Search PubMed.
- K. K. Diehn, H. Oh, R. Hashemipour, R. G. Weiss and S. R. Raghavan, Soft Matter, 2014, 10, 2632–2640 RSC.
- Polymer Blends Handbook, Springer Science & Business Media.
- J. E. Mark, Physical Properties of Polymers Handbook, Springer Science & Business Media, 2007 Search PubMed.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS PubMed.
- K.-H. Liao, S. Aoyama, A. A. Abdala and C. Macosko, Macromolecules, 2014, 47, 8311–8319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Transmission electron micrographs and electron diffraction patterns of graphene nanoplatelets for different graphene concentrations in 2-ethyl-1-hexanol (Fig. S1 and S2); edge view of graphene nanoplatelets (Fig. S3); steady-shear viscosity data at 50 °C (Fig. S4); creep data fitted with Maxwell–Jefferys model (Fig. S5) and stretched exponential function (Fig. S6); schematic of the viscoelastic Maxwell–Jeffreys model (Fig. S7). See DOI: 10.1039/c5ra13672e |
| This journal is © The Royal Society of Chemistry 2015 |