
Novel TiO2/graphene oxide functionalized with a cobalt complex for significant degradation of NOx and CO†
 a
and
a
and
Abstract
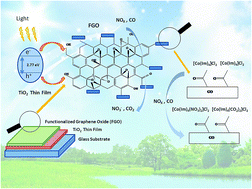
We are reporting a well-designed nanostructured composite, consisting of titanium dioxide (TiO2) and cobalt (Co) imidazole (Im) complex functionalized graphene oxide (FGO). The Co–Im complex was attached on the GO by covalent bonding of Im with the graphene functional groups. The films were characterized by XRD, XPS, TEM, UV-vis, FTIR and Raman spectroscopy in order to subsequently prove the evidence of hybridization of GO with the Co–Im complex. The band gap calculated for the TiO2 thin film was 3.10 eV, 2.96 eV for TiO2/GO and 2.776 eV for TiO2/FGO. Experimental results confirmed high-percentage deterioration of NOx (51%) and CO (46%). The photodegradation experiments revealed a significant photocatalytic character of TiO2, physical adsorption of GO, and the affinity of cobalt to undergo complex formation with pollutant gases. The TiO2/FGO composite increases air pollutant degradation by nearly three times that of the bare TiO2 thin film.
 Please wait while we load your content...
Please wait while we load your content...

