
Catalytic degradation of Acid Orange 7 with hydrogen peroxide using CoxOy-N/GAC catalysts in a bicarbonate aqueous solution
Abstract
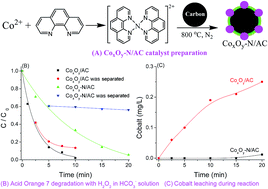
The cobalt-based heterogeneous catalysts CoxOy-N/GAC were prepared by pyrolysis of a cobalt–phenanthroline complex on granular active carbon (GAC) in a nitrogen atmosphere, and tested for the degradation of Acid Orange 7 using hydrogen peroxide as a benign oxidant in a bicarbonate aqueous solution. Characterization by X-ray diffraction, transmission electron microscopy, X-ray photoelectron spectroscopy, and electron spin resonance spectroscopy revealed the formation of a cobalt oxide nanoparticle shell with a metallic Co core on the surface of GAC; and the nitrogen substituted the selected carbon atoms during the pyrolysis process and bonded to cobalt. The catalysts were active for dye decolorization in an aqueous solution containing 10 mM H2O2 and 5 mM NaHCO3 at room temperature. They also presented good stability with nearly no loss of cobalt ions after the reaction, in comparison with the high leaching of Co (0.25 mg L−1) from the CoxOy/GAC catalyst without nitrogen. The production of intermediates, the formation of reactive radicals and the effect of HCO3− were also investigated to further explore the efficiency of the catalyst. This study can provide a promising way for the activation of the green oxidant H2O2 in a bicarbonate aqueous solution by heterogeneous cobalt catalysts for environmental remediation.
 Please wait while we load your content...
Please wait while we load your content...

