
In silico prediction of linear free energy relationship descriptors of neutral and ionic compounds†
Abstract
We present a prediction model for linear free energy relationship (LFER) descriptors – excess molar refraction (E), dipolarity/polarizability (S), hydrogen bonding acidity (A) & basicity (B), McGowan volume (V), and interaction of cations (J+) and anions (J−) – of both ionic and neutral compounds at the same scale. From computational calculations using density functional theory, a conductor screening model, and the OBPROP program in Turbomole, we obtained the following physicochemical sub-parameters for 992 molecules and atoms, polar surface area, molecular weight, volume, energy of van der Waals, sigma moments, molar refraction, and hydrogen-bond donor and acceptor abilities of a molecule or an atom. By making selective combinations of these sub-parameters – including also the number of rings, OH groups, and hydrogen atoms attached to nitrogen – we obtained prediction models for the LFER descriptors V, E, S, A, and B with reasonable accuracies, i.e. for a training set of compounds all R2 above 0.934. We validated the models by comparing calculated and experimentally determined LFER descriptors of a test set. Using the complete dataset, the following R2 and SE values were obtained: E (R2 = 0.949, SE = 0.136), S (R2 = 0.940, SE = 0.378), A (R2 = 0.936, SE = 0.148), B (R2 = 0.973, SE = 0.160), J+ (R2 = 0.816, SE = 0.351), and J− (R2 = 0.700, SE = 0.291). Furthermore, we demonstrated the applicability of the calculated LFER descriptors by predicting transfers of neutral and ionic compounds from water to propylene carbonate, sulfolane, and ethylene glycol with good accuracy. These results show that physicochemical properties of ionic and neutral compounds can be reliably predicted with identical LFER descriptors even for chemical entities that do not yet exist.
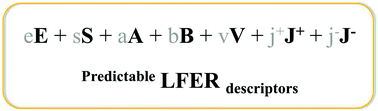
 Please wait while we load your content...
Please wait while we load your content...

