
Graphene oxide immobilized with ionic liquids: facile preparation and efficient catalysis for solvent-free cycloaddition of CO2 to propylene carbonate†
Abstract
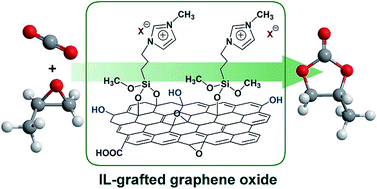
Functionalized imidazolium-based ionic liquids (ILs) with different halides (Cl, Br, and I) were successfully immobilized on the surface of graphene oxide materials by one step through covalent condensation between alcoholic hydroxyl groups of GO and alkoxyl groups of functionalized ILs. Several characterization including TG, Raman, AFM, FT-IR, and XPS techniques have been applied to characterize the physicochemical properties of the synthesized GO-[SmIm]X materials. In the solvent-free cycloaddition reactions of CO2 to propylene oxide, GO-[SmIm]I showed remarkably catalytic activity, affording a maximum yield of propylene carbonate as ca. 96%. The heterogeneous catalyst could be reused for at least four runs without any significant loss in activity, and demonstrated versatile catalysis for a wide range of substrates. A possible catalytic mechanism has been proposed, wherein epoxides were activated by the oxygen-containing groups of GO and the halide anions of the grafted ILs acted as key active species for the catalytic cycloaddition reactions.
 Please wait while we load your content...
Please wait while we load your content...

