
Synthesis, structure and electrochemical properties of lithium-rich cathode material Li1.2Mn0.6Ni0.2O2 microspheres
Juan Meng,
Shichao Zhang*,
Xin Wei,
Puheng Yang,
Shengbin Wang,
Jing Wang,
Honglei Li,
Yalan Xing and
Guanrao Liu
School of Materials Science and Engineering, Beihang University, Beijing 100191, PR China. E-mail: csc@buaa.edu.cn; Fax: +86 1082338148; Tel: +86 1082339319
First published on 14th September 2015
Abstract
Lithium-rich Li1.2Mn0.6Ni0.2O2 microspheres with a few mesopores have been successfully obtained. The results show that the Li1.2Mn0.6Ni0.2O2 material exhibits excellent cycling capability and rate performance. The microsphere morphology with a few mesopores could play a significant role in improving electrochemical performance.
1. Introduction
Lithium-ion batteries (LIBs) with high energy and power density are imperative to develop for electric vehicles (EV), hybrid electric vehicles (HEV) and plug-in hybrid electric vehicles (PHEV). For LIBs, it is cathode materials that determine energy density, cycle life and the cost. Thus the development of advanced cathode materials with less cost, higher capacity and more environmental friendliness is crucial in every essence. Layered lithium cobalt oxide (LiCoO2),1 spinel lithium manganese oxide (LiMn2O4),2 olivine lithium iron phosphate (LiFePO4)3 have been widely studied as cathode materials for several decades. However, these materials cannot extricate themselves from relatively low capacity: 140 mA h g−1 for LiCoO2, 148 mA h g−1 for LiMn2O4 and 170 mA h g−1 for LiFePO4.4,5 In addition, for Co-containing cathode materials, the sustainable development is restricted due to the limited resource, high cost and toxicity of cobalt element.6,7Recently, lithium-rich Li[Li1/3−2x/3Mn2/3−x/3Nix]O2, which is also denoted as xLi2MnO3·(1 − x)Li[Ni1/2Mn1/2]O2, cathode material has attracted much attention owing to its higher capacity (over 250 mA h g−1), lower cost, and less toxicity. It is considered as solid solution or integrated compound of layered Li2MnO3 phase (space group C2/m) and layered Li[Ni1/2Mn1/2]O2 (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m).8–15 However, Li[Li1/3−2x/3Mn2/3−x/3Nix]O2 have some intrinsic problems to be solved, like the relatively low cycling capability and poor rate performance.16–18
m).8–15 However, Li[Li1/3−2x/3Mn2/3−x/3Nix]O2 have some intrinsic problems to be solved, like the relatively low cycling capability and poor rate performance.16–18
It is well known that electrochemical properties are significantly influenced by the synthesis methods.8,10,15,19 As a result, various synthesis methods, such as co-precipitation method,11,19–21 sol–gel method22–24 and hydrothermal method,8,25–27 have been extensively exploited to improve electrochemical performance and overcome these mentioned drawbacks. Hydrothermal method is generally used for crystal growth control to obtain favorable morphology and enhanced correlative properties.
In this work, the precursor with mesoporous microspheres was successfully prepared by a facile hydrothermal method. Through calcined with a step procedure, lithium-rich cathode material Li1.2Mn0.6Ni0.2O2 (i.e., 0.5Li2MnO3·0.5Li[Ni1/2Mn1/2]O2) with a few mesopores were obtained, which is proved beneficial for electrochemical performances, especially for rate capability. The structure, morphology and electrochemical properties were thoroughly investigated. The cycling capability and rate performance are improved compared to some previous reports without mesoporous microspheres structure.28,29
2. Experimental
2.1 Synthesis of Li1.2Mn0.6Ni0.2O2
The precursor was prepared by a facile hydrothermal method. Stoichiometric amount of manganese(II) chloride, nickel(II) chloride with a molar ratio of 0.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 were dissolved in distilled water. Ammonium bicarbonate (NH4HCO3) was subsequently dropped in by stirring. The mixture was then transferred into a stainless steel autoclave and heated to 200 °C for 20 h in an oven to produce the precursor. The precursor was then dried at 120 °C and mixed with a certain amount of lithium carbonate. The dried mixture was preheated at 450 °C for 3 h and at 500 °C for 3 h, and then calcined at 700 °C for 1 h and at 900 °C for 6 h in the air. Ultimately, Li1.2Mn0.6Ni0.2O2 powder was obtained by cooling to room temperature in the furnace. All the raw materials were analytical purity grade and lithium carbonate was excess to compensate lithium loss during the calcination processes at high temperature.
:0.2 were dissolved in distilled water. Ammonium bicarbonate (NH4HCO3) was subsequently dropped in by stirring. The mixture was then transferred into a stainless steel autoclave and heated to 200 °C for 20 h in an oven to produce the precursor. The precursor was then dried at 120 °C and mixed with a certain amount of lithium carbonate. The dried mixture was preheated at 450 °C for 3 h and at 500 °C for 3 h, and then calcined at 700 °C for 1 h and at 900 °C for 6 h in the air. Ultimately, Li1.2Mn0.6Ni0.2O2 powder was obtained by cooling to room temperature in the furnace. All the raw materials were analytical purity grade and lithium carbonate was excess to compensate lithium loss during the calcination processes at high temperature.
2.2 Characterization and electrochemical tests of Li1.2Mn0.6Ni0.2O2
X-ray diffraction (XRD, Rigaku D/Max-2400, Japan) with Cu Kα radiation was used to identify the crystalline structure of materials. The XRD spectrum was collected in the range of 2θ value from 10° to 80°. The chemical stoichiometry was determined by Inductively Coupled Plasma-Atomic Emission Spectrometer (ICP-AES, PROFILE SPEC, Leeman). Scanning electron microscopy (SEM, Hitachi S-4800, Japan) and high-resolution transmission electron microscopy (TEM, Tecnai G2 F30, FEI) were engaged to observe the shape, size and distribution. Specific surface areas of the powders were measured by the multipoint Brunauer–Emmett–Teller (BET) method. The chemical valence state of all the elements were determined through X-ray photoelectron spectroscopy (XPS) by using ESCALAB250 (Thermo Fisher Scientific) with monochromatic Al Kα anode source with pass energy of 20 eV and energy step of 0.1 eV.Electrochemical properties were investigated by assembling half-cells in an argon-filled glove box (MB-10 G with TP170b/mono, MBRAUN) with lithium metal as counter and reference electrode. The electrode was prepared by spreading a mixture of 80 wt% active material, 10 wt% carbon black, and 10 wt% polyvinylidene fluoride (PVDF) with n-methyl-2-pyrrolidone (NMP) onto an Al foil current collector. The electrode was then dried at 120 °C for 10 h in a vacuum oven. The electrolyte was formed by 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC), dimethyl carbonate (DEC) and ethyl–methyl carbonate (DMC) (1:1:1 in volume). The charge–discharge measurements were galvanostatically carried out by using a battery test system (NEWARE BTS-610, Neware Technology Co., Ltd, China) in the voltage range of 2.0–4.8 V at room temperature. The current at 1C corresponds to 250 mA g−1. The cyclic voltammograms (CV) were tested by using an electrochemical station (CHI660a) at a scanning rate of 0.1 mV s−1 in the voltage range of 2.0–4.8 V. The electrochemical impedance spectra (EIS) measurements were carried out at frequencies from 100 kHz to 10 mHz with amplitude of 5 mV using an electrochemical station (CHI660a). All electrochemical measurements were conducted at room temperature.
3. Results and discussion
3.1 Material characterization
Fig.1 shows SEM images of the precursor (a), the intermediates (b–d) and final product (e and f). The precursor obtained by hydrothermal method displays uniform microspheres with a diameter of about 3 μm, as shown in Fig. 1a. The microsphere morphology is well maintained through a step procedure sintering from 400 °C to 900 °C, as shown in Fig. 1b–e. The microspheres almost retain the same diameter size (approximate 3 μm) before being calcined at 900 °C (Fig. 1a–d). The lithium-rich cathode material is obtained after being heated at 900 °C for 6 h and the diameter of microsphere is 6–7 μm, two times larger than that at previous state. The surface of microsphere turns obviously rough and the red region signed in Fig. 1e is magnified and shown in Fig. 1f. With the combination of Fig. 1e and f, the final product displays a secondary microsphere densely aggregated by nanoparticles of about 150 nm. The molecular formula of as-prepared lithium-rich cathode material obtained by ICP is Li1.256Mn0.595Ni0.182O2, listed in Table 1. | ||
| Fig. 1 SEM images of precursor (a) obtained by hydrothermal process, the intermediates preheated at 450 °C for 3 h (b), at 500 °C for 3 h (c) and calcined at 700 °C for 1 h (d), as-synthesized lithium-rich Li1.2Mn0.6Ni0.2O2 cathode material calcined at 900 °C for 6 h (e and f). | ||
| Element ratio in mole | Chemical formula | ||||
|---|---|---|---|---|---|
| Li | Mn | Ni | O | ||
| Designed | 1.200 | 0.600 | 0.200 | 2.000 | Li[Li0.2Mn0.6Ni0.2]O2 |
| Measured | 1.256 | 0.595 | 0.182 | 2.000 | Li1.256Mn0.595Ni0.182O2 |
The microspheres architecture of precursor (Fig. 2a) and Li1.2Mn0.6Ni0.2O2 powder (Fig. 2b–d) can also be clearly observed through TEM images. The HRTEM image (Fig. 2b) demonstrates that the distance of 0.47 nm agrees well with the (003) lattice spacing of the hexagonal Li[Ni1/2Mn1/2]O2 and the (001) for monoclinic Li2MnO3, indicating the formation of highly crystalline oxide.30 The shape and size are accordant with the results observed from the SEM images. Furthermore, some pores can be clearly observed in the microspheres of precursor. The microspheres of Li1.2Mn0.6Ni0.2O2 powder are larger and thicker than that of precursor.
 | ||
| Fig. 2 TEM images of precursor (a) and Li1.2Mn0.6Ni0.2O2 powder (b–d). | ||
Nitrogen adsorption–desorption isotherms and pore distribution curves (Fig. 3a and b) demonstrate that the precursor exhibits mesoporous microspheres and the BET surface area is 87.179 m2 g−1. A large amount of mesopores and large BET surface area are mainly attributed to the gas release from the decomposition of NH4HCO3, and the evolution of NH3 and CO2 generated the pores in the microstructures. However, compared with precursor, the as-prepared microspheres, with the BET surface area of 1.528 m2 g−1, own fewer mesopores with smaller pore size, which is probably due to high surface energy and high levels of agglomeration, the particles are unceasingly densely aggregating to form a larger microsphere during high temperature heating processes. The primary nanoparticles and mesopores could effectively shorten the Li+ migration length. Meanwhile, moderate specific surface area (1.528 m2 g−1) could refrain from side reactions during the charge–discharge process. This kind of structure and morphology is beneficial to the electrochemical performances.
 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms and pore distribution curves of precursor (a and b) and Li1.2Mn0.6Ni0.2O2 powder (c and d). | ||
Fig. 4 shows the XRD patterns of precursor (a), intermediate products (b–d) and Li1.2Mn0.6Ni0.2O2 (e). The precursor is obtained by hydrothermal process and the XRD peaks are attributed to Ni0.25Mn0.75CO3 (Fig. 4a). The XRD patterns of the phase transformation during the solid state reaction between Ni0.25Mn0.75CO3 and LiCO3 at 450 °C, 500 °C, 700 °C and 900 °C are shown in Fig. 4b–e respectively. The thermal decomposition of the transition metal carbonate precursor produces NiMnO3, Mn2O3 and CO2 when the temperature is more than 200 °C.31 Its decomposition reaction follows the scheme:
| 4Ni0.25Mn0.75CO3 → NiMnO3 + Mn2O3 + 3CO2↑ | (1) |
 | ||
| Fig. 4 XRD patterns of precursor (a), the intermediates preheated at 450 °C for 3 h (b), at 500 °C for 3 h (c) and calcined at 700 °C for 1 h (d), as-synthesized lithium-rich Li1.2Mn0.6Ni0.2O2 cathode material calcined at 900 °C for 6 h (e). | ||
Then the reaction between NiMnO3 and Li2CO3 takes place in the stoichiometric ratio (the total transition metals/Li = 1) to form the Rm phase (Fig. 4b–d), which follows the equation:
| NiMnO3 + Li2CO3 → 2LiNi0.5Mn0.5O2 + CO2↑ | (2) |
Further decomposition of the residual Li2CO3 engages at the temperature higher than 700 °C (Fig. 4e).
It can be observed from Fig. 4b–d that two major diffraction peaks at 18.64° and 44.54° (2θ, highlighted by blue lines) appear during the calcinations process. The two lines can be indexed to (003) and (104) peaks based on the Rm space group. In the meantime, the peaks at 21.3°, 30.5° and 31.9° (2θ, highlighted by red lines), corresponding to (110), (20![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) and (002) diffraction peaks of Li2CO3, fade during the heat-treatment from 450 to 900 °C.31 After high temperature treatment at 900 °C for 6 hours, as-synthesized lithium-rich Li1.2Mn0.6Ni0.2O2 cathode material is obtained and the XRD pattern is shown in Fig. 4e. All the diffraction peaks can be indexed to a hexagonal α-NaFeO2 structure with the Rm space group except for the weak peaks between 20° and 23°. These weak peaks identified as the (020) reflection are attributed to the super lattice structure of Li2MnO3 component (C2/m space group).32 Lattice parameters (a = 2.860(3) Å and c = 14.228(3) Å) are close to the previous reported value. The c/a value is 4.97 (>4.9), indicating a satisfactory crystalline layered structure.32 The clear splitting between the adjacent peaks of (006)/(102) and (108)/(110) confirms the highly ordered layered structure.33 The intensity ratio of (003)/(104) is used to denote the degree of cationic disorder in the Li-layers.32 The value of I(003)/I(104) is 1.377 (>1.2), reflecting a favorable layered structure without undesirable cationic disorder.
) and (002) diffraction peaks of Li2CO3, fade during the heat-treatment from 450 to 900 °C.31 After high temperature treatment at 900 °C for 6 hours, as-synthesized lithium-rich Li1.2Mn0.6Ni0.2O2 cathode material is obtained and the XRD pattern is shown in Fig. 4e. All the diffraction peaks can be indexed to a hexagonal α-NaFeO2 structure with the Rm space group except for the weak peaks between 20° and 23°. These weak peaks identified as the (020) reflection are attributed to the super lattice structure of Li2MnO3 component (C2/m space group).32 Lattice parameters (a = 2.860(3) Å and c = 14.228(3) Å) are close to the previous reported value. The c/a value is 4.97 (>4.9), indicating a satisfactory crystalline layered structure.32 The clear splitting between the adjacent peaks of (006)/(102) and (108)/(110) confirms the highly ordered layered structure.33 The intensity ratio of (003)/(104) is used to denote the degree of cationic disorder in the Li-layers.32 The value of I(003)/I(104) is 1.377 (>1.2), reflecting a favorable layered structure without undesirable cationic disorder.
XPS was carried out to ascertain chemical valence of the surface for Li1.2Mn0.6Ni0.2O2 powder. The spectra of Ni 2p, Mn 2p, O 1s and Li 1s are shown in Fig. 5. It can be detected that Ni2+ (854.4 eV) and Ni3+ (855.3 eV) have co-existed on the surface of cathode, as shown in Fig. 5a and b.24 There occurs oxidation reaction of partial amount of Ni2+ during the synthesis process, which is probably due to over-lithiation.17 According to Mn 2p (Fig. 5c and d) and Mn 3p spectra (Fig. 5f), Mn4+ is the majority constituent with respect to the peaks at 642.3 eV (Mn 2p3/2), 653.7 eV (Mn 2p1/2) and 50.3 eV (Mn 3p). The peaks at 641.8 eV (Mn 2p3/2) and 49.7 eV (Mn 3p) are attributed to Mn3+,24 indicating that a small amount of Mn3+ exists on the surface. The decrease of average valence for Mn may imply the formation of spinel structure. From Fig. 5e, the large peak of O 1s at 529.2 eV is assigned to the lattice oxygen. The small peak at 531.5 eV can probably be attributed to the residual Li2CO3.37
 | ||
| Fig. 5 Ni 2p (a and b), Mn 2p (c and d), O 1s (e) and Li 1s (f) XPS spectra of Li1.2Mn0.6Ni0.2O2 powder. | ||
3.2 Electrochemical properties
Fig. 6 displays the cyclic voltammetry measurements of Li1.2Mn0.6Ni0.2O2 electrode for the first 10 cycles. The oxidation and reduction peaks of the first cycle are different from those of the subsequent cycles, indicating a special charge and discharge mechanism in initial cycle, as is the character of Li-rich cathodes. In initial positive scanning (Fig. 6a), the oxidation peaks around 3.85 V and 4.14 V are ascribed to the oxidation of Ni2+/Ni3+ and Ni3+/Ni4+, respectively.12 The sharp oxidation peak at 4.69 V can be assigned to the irreversible oxidation of O2−, corresponding to electrochemical activation of Li2MnO3, which disappears in subsequent cycles.10,34–36 The reduction peaks at 3.83 V and 3.29 V are attributed to the reduction of Ni and Mn4+, respectively. The reduction of Mn4+ ions brings about extra lithium insertion, leading to improved discharge capacity in subsequent cycles. Although a small amount of Mn3+ is analyzed through the XPS spectra, oxidation peak of Mn3+ cannot be observed in the first oxidation cycle of CV curves, which probably indicates that only a small amount of Mn3+ exist on the surface. From 2nd to 10th cycles (Fig. 6b), the oxidation peaks of Mn3+ and Ni2+ appear at 3.3–3.4 V and 3.89–4.0 V, respectively.10 The oxidation peaks of Ni3+ shift to 4.37 V, suggesting that the electronic environment for high-valence nickel ions is affected by structural rearrangement which is originated from irreversible loss of oxygen in the initial cycle. The reduction peaks in the range of 4.3–3.6 V belong to the reduction of Ni4+ to Ni2+. The reduction peaks of Mn4+/Mn3+ (3.3–3.1 V) shift to the low voltage gradually with cycling, which is associated to a part of structure transformation from layer to spinel.37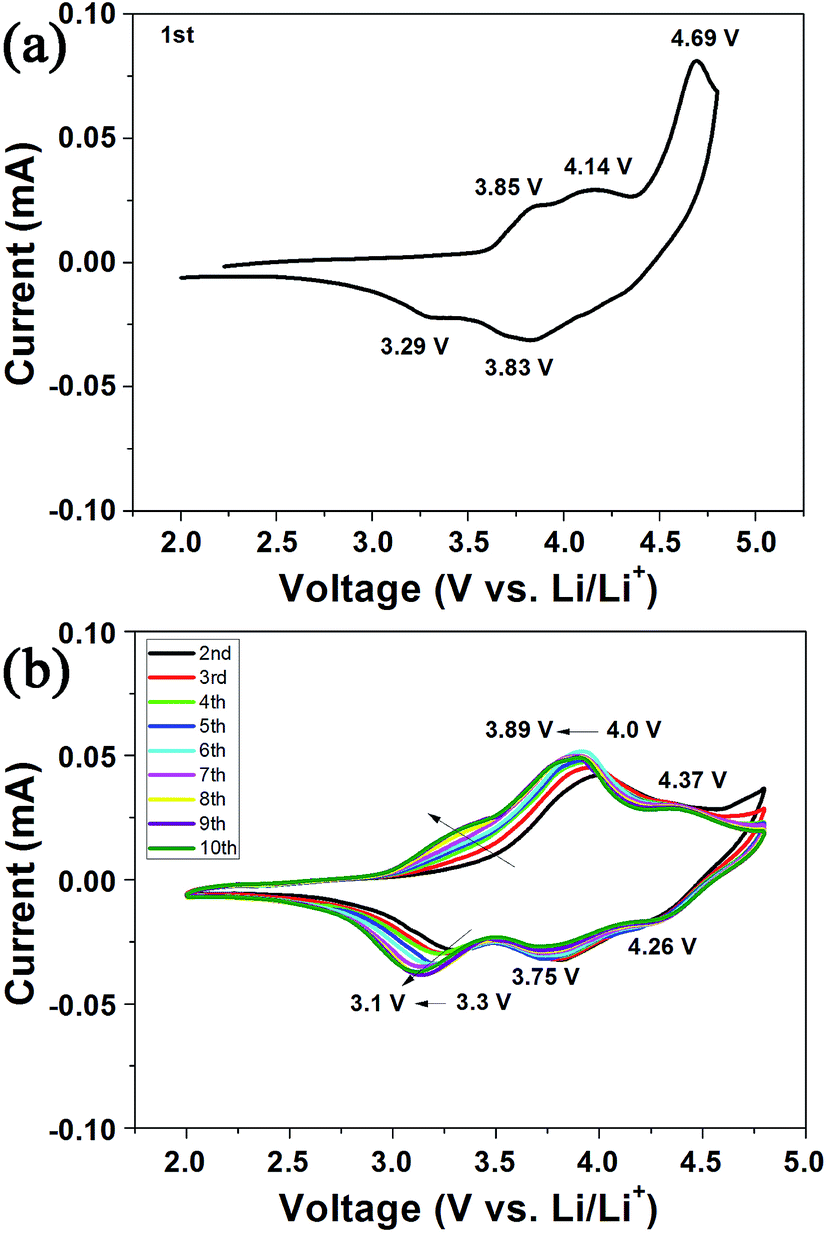 | ||
| Fig. 6 Cyclic voltammogram of Li1.2Mn0.6Ni0.2O2 electrode for the first 10 cycles. | ||
The charge–discharge profiles of Li1.2Mn0.6Ni0.2O2 electrode at 0.1C are shown in Fig. 7a. The initial charge curve shows two plateau located at 3.7–4.5 V and above 4.5 V, indicating two different Li-extraction processes during the first charge process.38 The low-voltage plateau is associated with the Li-extraction from the structure of space group Rm at 3.7–4.5 V, corresponding to the oxidation of Ni2+ to Ni4+.39 The second plateau (>4.5 V) is widely accepted as simultaneous oxygen release with Li-extraction from the layered Li2MnO3 lattice, which results in high irreversible capacity loss.40,41 The plateau situated at ∼4.5 V region no longer appears reversibly after the first cycle. The first charge and discharge capacity are respectively 392.0 and 256.4 mA h g−1 with a coulombic efficiency of 65.4%. The 2nd charge capacity drops to 257.7 mA h g−1, which is due to irreversible extraction of Li2O in the first charge process. The 2nd discharge capacity is 246.4 mA h g−1 and the coulombic efficiency is 95.6%. After 10 cycles, charge and discharge capacity are respectively 235.4 and 225.1 mA h g−1, corresponding to a coulombic efficiency of 95.6%. Fig. 7b shows the first discharge curves for Li1.2Mn0.6Ni0.2O2 electrode measured at 0.1C, 1C and 2C. The electrode delivers capacities of 256.4, 215.6 and 125.7 mA h g−1 at 0.1C, 1C and 2C, respectively. The initial discharge capacity decreases with the increase of C-rates, which is probably owing to the ascent of polarization for electrodes, and it could impact the lithium ion intercalation/deintercalation.
 | ||
| Fig. 7 Charge–discharge profiles at 0.1C (a) and initial discharge profiles at 0.1C, 1C, 2C (b) of Li1.2Mn0.6Ni0.2O2 electrode. | ||
Fig. 8a depicts the cycle performance of Li1.2Mn0.6Ni0.2O2 electrode for 100 cycles at different rates from 2 to 4.8 V.29 The discharge capacities of 0.1C and 1C gradually decrease with cycling. After 100 cycles, the discharge capacities retain 47.8% (122.6 mA h g−1) and 85.0% (183.3 mA h g−1) at 0.1C and 1C, respectively. The electrode has relatively low capacity in initial cycles at 2C. With the increase of the cycle number, the discharge capacity shows an increasing trend and rises to as much as 163.3 mA h g−1 after 100 cycles. It is probably attributed to gradually adequate activation of Li2MnO3. It can be observed that the capacity decreases with the increase of the C-rates and polarization. Since smaller rates bring about deeper charge and discharge reaction, quantities of lithium ions deinsert from their place, leading to many lithium vacancies. On the one hand, some nickel ions are likely to transport into lithium ion vacancies; on the other hand, vacancies may increase the chance of structural collapse in cycles that followed. So, lithium ions which can go back and insert into its position might be less than it should be. Therefore, the discharge capacity decreases more quickly at 0.1C than that at larger rates.
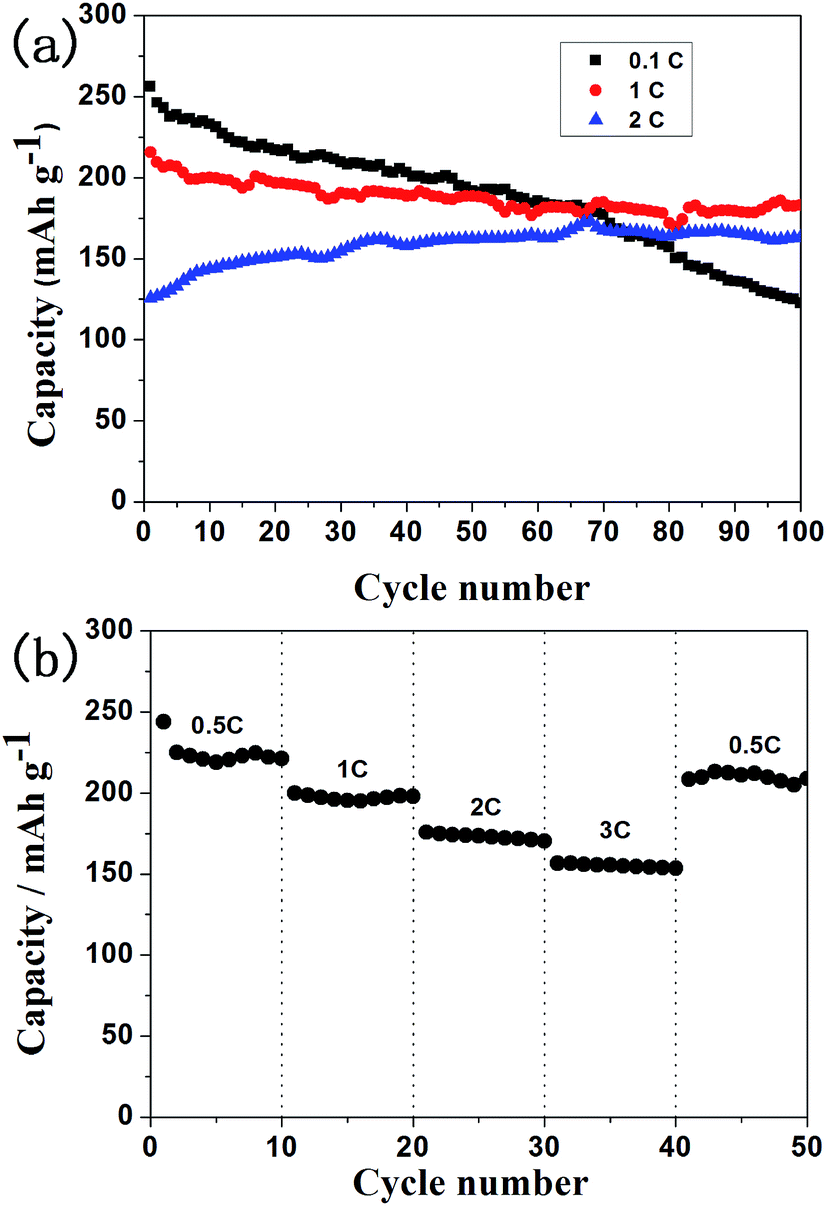 | ||
| Fig. 8 Cycle performance of Li1.2Mn0.6Ni0.2O2 electrode for 100 cycles at different rates (a) and its rate performance from 0.5C to 3C (b). | ||
Fig. 8b shows the rate performance of Li1.2Mn0.6Ni0.2O2 electrode from 0.5C to 3C. It can be seen that the electrode displays high capacity and excellent rate performance. The cathode delivers the average discharge capacities of 225 mA h g−1, 200 mA h g−1, 175.7 mA h g−1 and 156.8 mA h g−1 at 0.5C, 1C, 2C and 3C, respectively. About 92.7% (208.6 mA h g−1) of the initial discharge capacity can be recovered back at 0.5C.
To further investigate the electrochemical properties, electrochemical impedance spectroscopy (EIS) measurements were carried out. The cells were fully charged to 4.8 V and cycled at 1C after 1, 50 and 100 times before being tested and the Nyquist plots, relevant equivalent circuit for them are shown in Fig. 9. In the equivalent circuit, Rs mean the resistance for the transportation of Li+ through the interfacial film, Rct represents the charge transfer resistance, CPE refers to the constant phase-angle element, which depicts the nonideal characteristic of the double layer, Zw indicates the Warburg impedance relating to Li+ diffusion in the bulk material. There exists a semicircle at high frequency and a slope line at low frequency. The diameter of the semicircle is an indication of the resistance when Li+ diffuses through the interfacial film, i.e., the charge transfer resistance (Rct). The slope line is attributed to Li+ diffusion in the bulk material.6 A higher Rct value reflects larger electrode polarization, thus causing worse rate capability. The Rct value, which is subjected to large variation within extensive cycles, increases from 178.9 to 388.3 Ω from 1 cycle to 100 cycles. The relative low Rct value and the deviation also imply an improved electrochemical performance.
 | ||
| Fig. 9 EIS spectra and equivalent circuits of Li1.2Mn0.6Ni0.2O2 electrode. | ||
The excellent electrochemical properties could be ascribed to three aspects. On the one hand, the primary nanoparticles and a few mesopores could effectively shorten the Li+ migration length. On the other hand, relatively small specific surface area could refrain from partial side reaction during the charge–discharge process. In addition, a few spinel structures at the surface region originated from the small amount of Mn3+ is helpful to improve rate capability, which is due to the faster diffusion of Li+.17,24
4. Conclusions
In summary, lithium-rich layered cathode material Li1.2Mn0.6Ni0.2O2 has been successfully synthesized and thoroughly investigated. The cathode material owns a desired crystalline layered structure and secondary microsphere morphology with a few mesopores. The primary nanoparticles and mesopores could effectively shorten the Li+ migration length. Meanwhile, relatively small specific surface area could refrain from side reactions during the charge–discharge process. The cathode material exhibits excellent cyclic capability and rate performance. It retains 85.0% of initial discharge capacity after 100 cycles at 1C. After the battery cycles at the increasing rate from 0.5C to 3C, about 92.7% (208.6 mA h g−1) of the initial discharge capacity is recovered back at 0.5C. And it holds such desirable rate capability as to more than 90% of the initial capacity could be recovered after designed cycles. Thus, the hydrothermal method applied in this work has provided a new approach to synthesize a functional structure for lithium-rich layered cathode materials.Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program) (2013CB934001), National Natural Science Foundation of China (51274017), National 863 Program of China (2013AA050904), International S&T Cooperation Program of China (2012DFR60530), Shanghai Academy of Space Technology (SAST201467).Notes and references
- X. Y. Qiu, Q. C. Zhuang, Q. Q. Zhang, R. Cao, P. Z. Ying, Y. H. Qiang and S. G. Sun, Phys. Chem. Chem. Phys., 2012, 14, 2617–2630 RSC.
- X. D. Xiang, Z. Fu and W. S. Li, J. Solid State Electrochem., 2013, 17, 1201–1206 CrossRef CAS.
- H. B. Shu, X. Y. Wang, Q. Wu, B. N. Hu, X. K. Yang, Q. L. Wei, Q. Q. Liang, Y. S. Bai, M. Zhou, C. Wu, M. F. Chen, A. W. Wang and L. L. Jiang, J. Power Sources, 2013, 237, 149–155 CrossRef CAS PubMed.
- H. Koga, L. Croguennec, M. Menetrier, P. Mannessiez, F. Weill, C. Delmas and S. Belin, J. Phys. Chem. C, 2014, 118, 5700–5709 CAS.
- Q. Q. Zou, G. N. Zhu and Y. Y. Xia, J. Power Sources, 2012, 206, 222–229 CrossRef CAS PubMed.
- F. Y. Cheng, J. Liang, Z. L. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- G.-Z. Wei, X. Lu, F.-S. Ke, L. Huang, J.-T. Li, Z.-X. Wang, Z.-Y. Zhou and S.-G. Sun, Adv. Mater., 2010, 22, 4364–4367 CrossRef CAS PubMed.
- G. Singh, R. Thomas, A. Kumar and R. S. Katiyar, J. Electrochem. Soc., 2012, 159, A410–A420 CrossRef CAS PubMed.
- X. Xiang, X. Li and W. Li, J. Power Sources, 2013, 230, 89–95 CrossRef CAS PubMed.
- Z. H. Lu, D. D. MacNeil and J. R. Dahn, ECS Solid State Lett., 2001, 4, A191–A194 CrossRef CAS PubMed.
- Z. H. Lu, L. Y. Beaulieu, R. A. Donaberger, C. L. Thomas and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A778–A791 CrossRef CAS PubMed.
- M. M. Thackeray, S. H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- C.-C. Wang and A. Manthiram, J. Mater. Chem. A, 2013, 1, 10209–10217 CAS.
- C.-C. Wang, K. A. Jarvis, P. J. Ferreira and A. Manthiram, Chem. Mater., 2013, 25, 3267–3275 CrossRef CAS.
- C. S. Johnson, N. Li, C. Lefief and M. M. Thackeray, Electrochem. Commun., 2007, 9, 787–795 CrossRef CAS PubMed.
- A. Abouimrane, O. C. Compton, H. Deng, I. Belharouak, D. A. Dikin, S. T. Nguyen and K. Amine, ECS Solid State Lett., 2011, 14, A126–A129 CrossRef CAS PubMed.
- A. Boulineau, L. Simonin, J.-F. Colin, C. Bourbon and S. Patoux, Nano Lett., 2013, 13, 3857–3863 CrossRef CAS PubMed.
- C. R. Fell, K. J. Carroll, M. F. Chi and Y. S. Meng, J. Electrochem. Soc., 2010, 157, A1202–A1211 CrossRef CAS PubMed.
- X. D. Xiang and W. S. Li, Electrochim. Acta, 2014, 133, 422–427 CrossRef CAS PubMed.
- A. R. Armstrong, M. Holzapfel, P. Novak, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- J. Shojan, C. V. Rao, L. Torres, G. Singh and R. S. Katiyar, Mater. Lett., 2013, 104, 57–60 CrossRef CAS PubMed.
- S. H. Kang and K. Amine, J. Power Sources, 2003, 124, 533–537 CrossRef CAS.
- B. H. Song, C. F. Zhou, Y. Chen, Z. W. Liu, M. O. Lai, J. M. Xue and L. Lu, RSC Adv., 2014, 4, 44244–44252 RSC.
- X. Wei, S. Zhang, Z. Du, P. Yang, J. Wang and Y. Ren, Electrochim. Acta, 2013, 107, 549–554 CrossRef CAS PubMed.
- L. Zhang, B. Wu, N. Li, D. Mu, C. Zhang and F. Wu, J. Power Sources, 2013, 240, 644–652 CrossRef CAS PubMed.
- F. Fu, Y. Y. Huang, P. Wu, Y. K. Bu, Y. B. Wang and J. N. Yao, J. Alloys Compd., 2015, 618, 673–678 CrossRef CAS PubMed.
- P. Z. Ying, X. Y. Qiu, Q. Q. Zhang and Q. C. Zhuang, Ionics, 2015, 21, 657–665 CrossRef CAS.
- X. D. Xiang and W. S. Li, Electrochim. Acta, 2014, 127, 259–265 CrossRef CAS PubMed.
- S. J. Shi, Z. R. Lou, T. F. Xia, X. L. Wang, C. D. Gu and J. P. Tu, J. Power Sources, 2014, 257, 198–204 CrossRef CAS PubMed.
- D. P. Wang, I. Belharouak, X. F. Zhang, Y. Ren, G. Meng and C. M. Wang, J. Electrochem. Soc., 2014, 161, A1–A5 CrossRef CAS PubMed.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- J. Lin, D. Mu, Y. Jin, B. Wu, Y. Ma and F. Wu, J. Power Sources, 2013, 230, 76–80 CrossRef CAS PubMed.
- D. Wang, Y. Huang, Z. Huo and L. Chen, Electrochim. Acta, 2013, 107, 461–466 CrossRef CAS PubMed.
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095–6106 CrossRef CAS.
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176–184 CrossRef CAS.
- X. L. Yuan, Q. J. Xu, C. Wang, X. N. Liu, H. M. Liu and Y. Y. Xia, J. Power Sources, 2015, 279, 157–164 CrossRef CAS PubMed.
- J. S. Kim, C. S. Johnson, J. T. Vaughey, M. M. Thackeray and S. A. Hackney, Chem. Mater., 2004, 16, 1996–2006 CrossRef CAS.
- C. S. Johnson, J. S. Kim, C. Lefief, N. Li, J. T. Vaughey and M. M. Thackeray, Electrochem. Commun., 2004, 6, 1085–1091 CrossRef CAS PubMed.
- Z. H. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef CAS PubMed.
- S. J. Shi, J. P. Tu, Y. Y. Tang, Y. Q. Zhang, X. L. Wang and C. D. Gu, J. Power Sources, 2013, 240, 140–148 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |