
Abstract
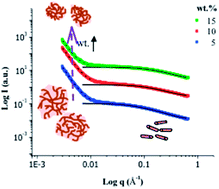
Recently, there has been a growing interest in developing value added uses for lignin, including the utilization of lignins as a precursor for carbon materials. Proper understanding of the association behavior of lignins during solution processing provides important structural information that is needed to rationally optimize the use of lignins in industry in a range of value added applications. In these experiments, we follow the assembly of lignin molecules from a variety of sources in dimethyl sulfoxide, a good solvent for lignins, using small angle neutron scattering. In order to mimic industrial processing conditions, concentrations of lignins were kept above the overlap concentration. At small length scales, short lignin segments with ∼4–10 monolignol units associate to form rigid rod-like/cylindrical building blocks, where the number of repeat units in a cylindrical segment decreases with increasing lignin concentration. These cylindrical building blocks associate to form aggregates with low cross-linking densities and a random coil or network like structures from highly branched lignin structures. The degree of branching of the base lignin molecule, which varies with source, plays a crucial role in determining their association behavior. The overall sizes of the aggregates decrease with increasing concentration at low cross-linking densities, whereas the opposite trend is observed for highly branched lignins.
 Please wait while we load your content...
Please wait while we load your content...

