
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of endoperoxides by domino reactions of ketones and molecular oxygen†‡
L.
Novkovic
a,
M.
Trmcic
a,
M.
Rodic
 b,
F.
Bihelovic
c,
M.
Zlatar
d,
R.
Matovic
d and
R. N.
Saicic
*c
b,
F.
Bihelovic
c,
M.
Zlatar
d,
R.
Matovic
d and
R. N.
Saicic
*c
aInnovative Centre of the Faculty of Chemistry, University of Belgrade, Serbia
bDepartment of Chemistry, University of Novi Sad, Novi Sad, Serbia
cFaculty of Chemistry, University of Belgrade, Studentski trg 16, POB 51, 11158 Belgrade 118, Serbia. E-mail: rsaicic@chem.bg.ac.rs
dICTM Center for Chemistry, Njegoseva 12, Belgrade, Serbia
First published on 13th November 2015
Abstract
Domino reactions of ketones with molecular oxygen in the presence of potassium hydroxide and potassium t-butoxide afford cyclic hydroperoxy acetals (3,5-dihydroxy-1,2-dioxanes). Mixed endoperoxides can also be obtained in a three-component reaction of two ketones and oxygen.
Introduction
Endoperoxides are important pharmacophores in a number of natural and synthetic, biologically active compounds. Although best known as potent antimalarials,1 cyclic peroxides also exhibit a range of activities which encompasses antitripanosomal,2 antifungal, antiviral and anticancer activity.3,4 Whereas tetraoxanes and trioxanes are important units of synthetic bioactive peroxides, 1,2-dioxanes and dioxenes are the most frequent constituents of naturally occuring endoperoxides. Therefore, synthetic chemists have invested considerable efforts in developing synthetic approaches to this class of compounds.5Results and discussion
During the course of a synthetic study of platensimycin, we exposed methyl cyclopentyl ketone 1 to the enolization conditions under a presumed argon atmosphere, aiming to perform a Michael addition with the thermodynamic enolate. Surprisingly, spectral data of the isolated compound did not match the expected product, but indicated a dimeric structure with a higher oxidation level. Detailed analysis of spectra indicated the product structure 2 (Scheme 1). Stereochemical assignment of the compound 2, as deduced from NOESY spectrum, was confirmed by the results of X-ray diffraction structural analysis, as represented in Fig. 1. As it was clear that the product arose from the reaction of ketone enolate with molecular oxygen, the reaction was performed under an oxygen atmosphere, in order to improve the yield; this time, surprisingly, endoperoxide 2 was not observed. The optimization of the reaction conditions involved variations of the base, solvent, atmosphere and the reaction temperature. We found that the highest yield of endoperoxide 2 could be obtained when the reaction was performed in THF, using a mixture of KOt-Bu and KOH as a base, under an argon atmosphere where oxygen was present only in low concentration; under these conditions, peroxide 2 could be isolated in 48% yield as a crystalline, analytically pure compound (Scheme 1, example 1). Cyclopent-3-enyl methyl ketone 3 behaved similarly, affording endoperoxide 4 in 43% yield (Scheme 1, example 2). | ||
| Scheme 1 Formation of 1,2-dioxanes 3 and 4 in the base-catalyzed reactions of ketones 1 and 2 with oxygen. | ||
 | ||
| Fig. 1 X-ray diffraction analysis of compound 2. | ||
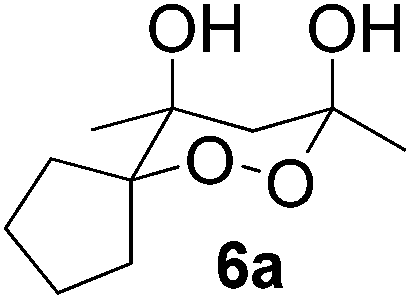
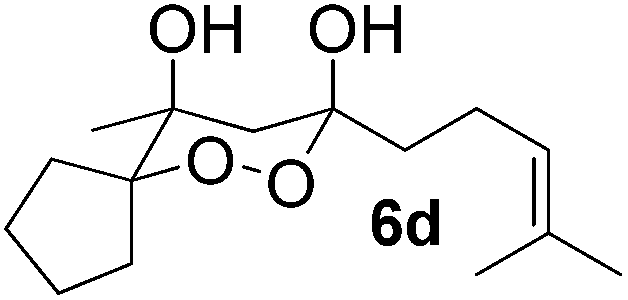
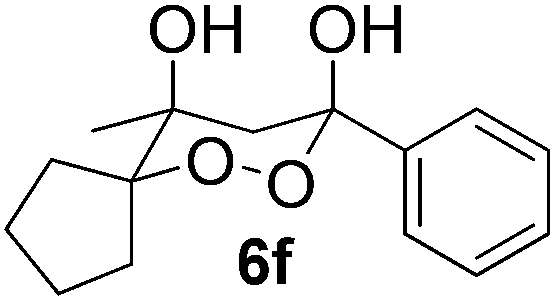
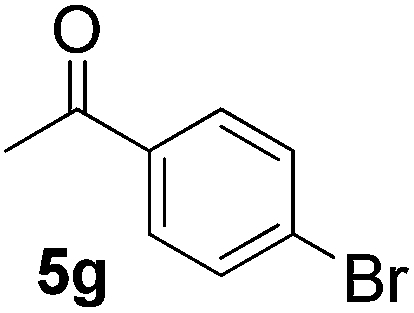
In subsequent experiments, attempts were made to accomplish cross-cyclotrimerization of two different ketones with oxygen. Indeed, the reaction of methyl cyclopentyl ketone and acetone afforded peroxide 6a in 64% yield (Table 1, entry 1). Several other ketones (5) were also submitted to the cross-cyclotrimerization sequence with methyl cyclopentyl ketone 1 and oxygen, to give the desired peroxide products (mostly as crystalline compounds) in modest to fair yields. In some cases the products (6) contained variable amounts of the side product of homo-trimerization (2) (entries 2, 3, 4 and 5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reactant (MeC(O)R1) | Product | T (°C) | Time (h) | Yielda (%) |
a Unless otherwise stated, the yields refer to the highest isolated yields of recrystallized, analytically pure compounds.
b Yield determined from 1H NMR spectrum.
c
6b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 = 2.9:1.
d
6c:2 = 17:1.
e
6d:2 = 5:1.
f
6e:2 = 6:1.
g
6f:2 = 9:1. :2 = 2.9:1.
d
6c:2 = 17:1.
e
6d:2 = 5:1.
f
6e:2 = 6:1.
g
6f:2 = 9:1.
|
|||||
| 1 |

|
|
−20 | 5 | 64 |
| 2 |

|

|
−20 | 2 | 38b,c |
| 3 |

|

|
−40 | 3.5 | 43 |
| 56b,d | |||||
| 4 |

|
|
−40 | 2 | 42b,e |
| 5 |

|

|
−40 | 5 | 39 |
| 59b,f | |||||
| 6 |

|
|
−40 | 6 | 31 |
| 54b,g | |||||
| 7 |
|

|
−40 | 6 | 67 |
| 8 |

|

|
−40 | 4 | 28 |
However, the preponderance of the cross-products (6) is of note, given the fact that ketones were used in equimolar amounts. The results of these experiments are represented in Table 1. All products were obtained as single stereoisomers, with both hydroxy groups assuming axial positions (Fig. 2).
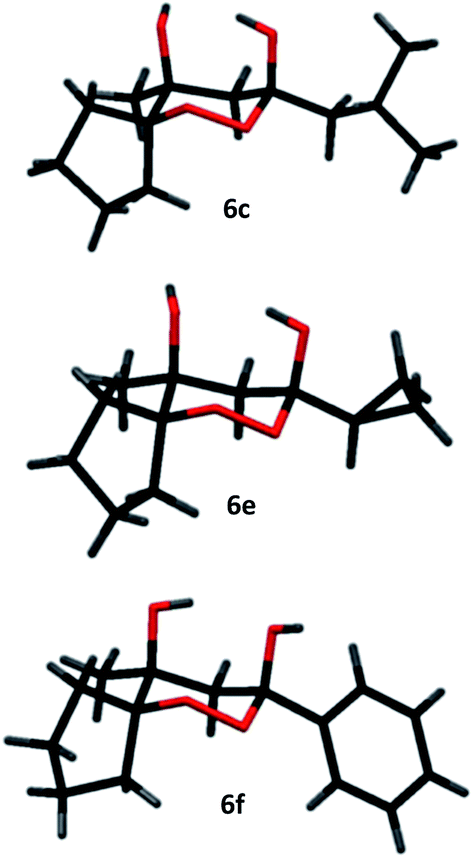 | ||
| Fig. 2 Crystal structures of compounds 6c, 6e and 6f. | ||
A literature search revealed that Barton and collaborators encountered this reaction while inspecting more closely autooxidation of isobutyrophenone.6 However, no subsequent report on this reaction ensued, although “the phenomenon certainly deserves further study”.7
The proposed reaction mechanism is represented in Scheme 2. It involves the oxidation of methyl cyclopentyl ketone enolate 7 with molecular oxygen to give hydroperoxide 8, which then acts as the electrophile in subsequent aldol addition of enolate 9. Selectivity in cross-cyclotrimerization can be explained by the well known propensity of methyl cyclopentyl ketone for the formation of a thermodynamic enolate 7,8 which undergoes the reaction with molecular oxygen.9 This reaction could proceed either as a direct reaction of the enolate with triplet oxygen, or via a cage radical pair mechanism; the intermediacy of free radical species can be ruled out, as the addition of BHT did not inhibit the reaction. The hydroperoxide thus formed would then act as the acceptor (probably activated by intramolecular hydrogen bond) in the cross aldol addition. Surprisingly, this mechanistic pathway was not corroborated with experimental evidence. Hydroperoxide 8 (represented in Scheme 2 in the form of the corresponding potassium salt) could be separately prepared in 71% yield, when the reaction of methyl cyclopentyl ketone with KOt-Bu was performed under an oxygen atmosphere (this reaction is also unaffected by the presence of BHT).10 However, a mixture of hydroperoxide 8 and methyl cyclopentyl ketone 1 did not produce observable amounts of endoperoxide 2 when exposed to the action of KOt-Bu.
 | ||
| Scheme 2 Proposed reaction mechanism. | ||
To clarify the relationship between enolate stability and the course of cross cyclotrimerization, and to explain the specific reactivity of methyl cyclopent(en)yl ketone(s) 1 and 3, dispersion corrected density functional theory calculations (DFT) were performed. Free energy changes for the t-BuO− mediated formation of more substituted enolates in THF solution have been calculated at BP86-D3/TZP level of theory (COSMO) with two methods. (The first one is based on direct calculation of thermodynamic properties in THF within the COSMO model,11 while in the second, indirect method, the gas-phase free energies of reactants and products are corrected by solvation energies and energies due to the geometry relaxation of a molecule in the solvent,12 see Computational details Section for details). The results of both, direct and indirect method, are consistent, and Gibbs free energies for the formation of more substituted enolates, corresponding to all aliphatic ketones used in cross trimerization reactions (1, 3, 5a–e) are listed in Table 2.14 It can be seen that methyl cyclopentyl ketone 1 and methyl cyclopentenyl ketone 3 have the most stable thermodynamic enolates. This indicates that methyl cyclopentenyl ketone 3 should also act as the acceptor in the cross cyclotrimerization reaction. Indeed, when 3 was submitted to the cyclotrimerization conditions in the presence of methyl isobutyl ketone 5c, endoperoxide 10 was obtained in 37% yield (Scheme 3, example 1). With these results in hand, we searched for some other ketone structures that would give rise to stable thermodynamic enolates and hence be good partners for the cross cyclotrimerization. Calculations showed that indanyl methyl ketone 11 should be such a compound (Table 2), and indeed, its' cyclotrimerization with acetone provided endoperoxide 12 in 77% yield (Scheme 3, example 2).
|
|
||
|---|---|---|
| Ketone | ΔG (kcal mol−1) | |
| Method I | Method II | |
| 1 | −3.92 | −3.99 |
| 3 | −4.08 | −4.48 |
| 5a | −3.34 | −3.05 |
| 5b | −3.72 | −3.14 |
| 5c | −3.13 | −2.46 |
| 5d | −3.81 | −3.54 |
| 5e | 7.04 | 7.32 |
| 11 | −5.45 | −5.40 |

 | ||
| Scheme 3 Reactions of ketones 3 and 11, as predicted by calculations. | ||
Some ketones did not participate in the cross-cyclotrimerization; these include progesterone, α-ionone, and 2-acetylnorbornene, inter alia. Therefore, we searched for a method to introduce the endoperoxide structural unit into various structures, not directly obtainable by the reaction. To this aim, cross-cyclotrimerisation was performed with acetylstyrene 13. Ozonolysis of the product 14 provided aldehyde 15, which could be submitted to further synthetic transformations, as examplified by the organometallic addition represented in Scheme 4, which gave peroxy-triol 16. In this way, various structural entities could be linked to the peroxide moiety, via nucleophilic addition.
 | ||
| Scheme 4 Linking the endoperoxide structural unit to other structures via organometallic addition. | ||
Conclusions
The new method for the synthesis of endoperoxides (1,2-dioxanes) is described, which relies on the base-catalyzed cyclotrimerization of two ketones with molecular oxygen. The domino reaction involves the oxidation of 1-cyclopentylethanone with molecular oxygen, cross-aldol addition and cyclization. The product of the reaction with acetylstyrene is amenable to further synthetic transformations, which allows for the linkage of the endoperoxide structural unit with other molecules.Experimental
General experimental
All chromatographic separations were performed on silica, 10–18, 60A, ICN Biomedicals. Standard techniques were used for the purification of reagents and solvents. NMR spectra were recorded on a Bruker Avance III 500. Chemical shifts are expressed in ppm (δ) using tetramethylsilane as internal standard. IR spectra were recorded on a Nicolet 6700 FT instrument, and are expressed in cm−1. Mass spectra were obtained on Agilent technologies 6210 TOF LC/MS instrument (LC: series 1200). Melting points were determined on an electrothermal apparatus and are corrected. Microanalyses were performed using the Vario EL III instrument CHNOS Elementar Analyzer, Elementar Analysensysteme GmbH, Hanau, Germany. Diffraction data were collected on an Oxford Diffraction KM4 four-circle goniometer equipped with Sapphire CCD detector.:1 ratio, as determined from the integrals of OH signals in the 1H NMR spectrum. This sample contained a small amount of an unidentified impurity which could not be separated by column chromatography. 1H NMR (500 MHz, CDCl3) δ 3.88 (d, J = 1.3 Hz, 1H, OH), 3.56 (s, 1H, OH), 2.26–2.16 (m, 1H), 2.03–1.92 (m, 1H), 1.91–1.43 (m, 7H), 1.79 (d, J = 14.0 Hz, 1H), 1.72 (dd, J = 14.0 and 1.2 Hz, 1H), 1.19 (s, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.96 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 103.3 (C), 96.3 (C), 71.0 (C), 37.7 (CH2), 36.3 (CH), 32.1 (CH2), 31.4 (CH2), 25.8 (CH2), 24.8 (CH2), 24.3 (CH3), 16.5 (CH3), 15.8 (CH3). IR (film) νmax: 3365, 3443, 3404, 2960, 2872, 1730, 1659, 1453, 1382, 1327. HRMS (ESI-TOF high acc.) calcd for C12H22O4 (M + NH4+): 248.1856, found: 248.1848.
:1 ratio, as determined from the integrals of OH signals in the 1H NMR spectrum. Crystallization from petroleum-ether/ethyl acetate gave white crystals (43%), mp 64–65 °C. 1H NMR (500 MHz, CDCl3) δ 4.19 (d, J = 1.0 Hz, OH), 3.42 (s, 1H, OH), 2.26–2.18 (m, 1H), 1.99–1.72 (m, 4H), 1.84 (d, J = 14.0 Hz, 1H), 1.75 (dd, J = 13.9 and 1 Hz, 1H), 1.70–1.39 (m, 6H), 1.18 (s, 3H), 0.98 (d, J = 9.1 Hz, 3H), 0.96 (d, J = 9.1 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 102.3 (C), 96.2 (C), 71.2 (C), 47.8 (CH2), 41.1 (CH2), 32.3 (CH2), 31.4 (CH2), 25.9 (CH2), 24.8 (CH2), 24.3 (CH3), 24.2 (CH3), 23.9 (CH3), 23.2 (CH). IR (film) νmax: 3412, 2957, 2871, 1711, 1454, 1380, 1297. HRMS (ESI-TOF high acc.) calcd for C15H26O4 (M + NH4+): 288.2169, found: 288.2161.
:1 ratio, as determined from the integrals of OH signals in the 1H NMR spectrum. 1H NMR (500 MHz, CDCl3) δ 5.09 (t, J = 7.0 Hz, 1H), 4.43 (s, 1H, OH), 3.69 (s, 1H, OH), 2.28–2.03 (m, 3H), 2.03–1.91 (m, 1H), 1.91–1.42 (m, 10H), 1.68 (s, 3H), 1.62 (s, 3H), 1.18 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 132.6 (C), 123.3 (CH), 101.7 (C), 96.4 (C), 71.0 (C), 40.6 (CH2), 38.9 (CH2), 32.1 (CH2), 31.4 (CH2), 25.8 (CH2), 25.6 (CH3), 24.8 (CH2), 24.1 (CH3), 21.2 (CH2), 17.6 (CH3). IR (film) νmax: 3432, 3374, 2958, 2931, 2869, 1666, 1450, 1449, 1379. HRMS (ESI-TOF high acc.) calcd for C15H26O4 (M + NH4+): 288.2169, found: 288.2161.
:1 ratio, as determined from the integrals of OH signals in the 1H NMR spectrum. Crystallization from petroleum-ether/ethyl acetate gave white crystals (39%), mp 106–107 °C. 1H NMR (500 MHz, CDCl3) δ 4.04 (s, 1H, OH), 3.42 (s, 1H, OH), 2.30–2.19 (m, 1H), 2.00–1.90 (m, 1H), 1.90–1.82 (m, 1H), 1.86 (s, 2H), 1.82–1.71 (m, 1H), 1.72–1.52 (m, 3H), 1.43–1.43 (m, 1H), 1.19 (s, 3H), 0.97 (tt, J = 8.4 and 5.3 Hz, 1H), 0.66–0.58 (m, 1H), 0.55–0.38 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 100.5 (C), 96.3 (C), 71.3 (C), 41.5 (CH2), 32.1 (CH2), 31.3 (CH2), 25.7 (CH2), 24.7 (CH2), 24.2 (CH3), 18.1 (CH), 0.7 (CH2), 0.1 (CH2). IR (film) νmax: 3424, 3362, 2960, 2872, 1659, 1636, 1471, 1381, 1322. HRMS (ESI-TOF high acc.) calcd for C12H20O4 (M + H+): 229.1434, found: 229.1431. Microanalysis: calcd for C12H20O4: C 63.14; H 8.83%; found: C 62.93; H 8.82.
:1 ratio, as determined from the integrals of OH signals in the 1H NMR spectrum. Crystallization from petroleum-ether/ethyl acetate gave white crystals (31%), mp (dec.) 125 °C. 1H NMR (500 MHz, CDCl3) δ 7.56–7.50 (m, 2H), 7.42–7.32 (m, 3H), 4.54 (s, 1H, OH), 3.60 (s, 1H, OH), 2.48–2.40 (m, 1H), 2.06 (s, 2H), 2.02 (dd, J = 14.8 and 7.6 Hz, 1H), 1.99–1.91 (m, 1H), 1.89–1.78 (m, 1H), 1.77–1.52 (m, 4H), 1.21 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 141.0 (C), 128.9 (CH), 128.4 (CH), 125.4 (CH), 101.1 (C), 96.5 (C), 71.3 (C), 43.7 (CH2), 32.4 (CH2), 31.5 (CH2), 25.9 (CH2), 24.9 (CH2), 24.1 (CH3). IR (film) νmax: 3357, 3139, 2976, 2933, 2867, 1707, 1657, 1632, 1492, 1449, 1431, 1381. HRMS (ESI-TOF high acc.) calcd for C15H20O4 (M + NH4+): 282.1700, found: 282.1697. Microanalysis: calcd for C12H20O4: C 68.16; H 7.63%; found: C 68.55; H 7.54.
:1, PAA) indicated that nearly all of the substrate was converted and the product was starting to decay. Ice-cold sat. NaHCO3 (3 mL) was added, layers were separated, pH of the aqueous layer was adjusted to 3–4 using conc. H3PO4 (∼0.5 mL), diluted with water and extracted with diethyl ether (2 × 10 mL). The combined etheral extract was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to afford the crude product, which was purified by column chromatography (SiO2; eluent: petroleum-ether/ethyl acetate = 6:1). Pure hydroperoxyde 8 (275.8 mg, 71%) was isolated as a colorless liquid. 1H NMR (500 MHz, CDCl3) δ 9.51 (s, 1H, OOH), 2.33 (s, 3H), 2.03–1.85 (m, 4H), 1.83–1.64 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 211.7 (C), 99.0 (C), 33.3 (CH2), 25.2 (CH3), 25.0 (CH2). IR (film) νmax: 3385, 2961, 2874, 1707, 1435, 1358. HRMS (ESI-TOF high acc.) calcd for C7H12O3 (M + NH4+): 162.1125, found: 162.1120.
Computational details
All the calculations have been carried out with the Amsterdam Density Functional program package, ADF2013.01.15 An all electron Triple-zeta Slater-type orbitals (STO) plus one polarization function (TZP) basis set has been used for all atoms. Solvent effects in THF solution are described with the COSMO model16 as implemented in ADF.17 Geometry optimizations of all investigated molecules, both in the gas phase and in solution, were performed using general gradient functional consisting of Becke's exchange18 and Perdew's correlation19 with Grimme's third-generation dispersion energy correction20 and Becke–Johnson dumping,21i.e. BP86-D3 functional, with Becke's integration grid of good quality.22 Analytical harmonic frequencies23 were calculated at the same level of theory, in order to ascertain that all the optimized structures correspond to the minima on the potential energy surface. In addition, vibrational analysis have been used to evaluate zero point effects, entropic and thermal corrections to the Gibbs free energy. Gibbs free energy changes for a reaction of a ketone with tert-butoxide, forming corresponding enolate and tert-butanol in THF solution, have been calculated with two methods. The two methods are employed because accurate description of reactions in solutions are still not at the same high levels as those in the gas phase, and there are debates in the literature concerning the free energy calculations with implicit solvation methods.24 The first method is direct calculation of thermodynamic properties within the COSMO model. COSMO model takes effectively into account cavitation, internal energy and entropy effects of the solvent, and therefore can yield an estimate of the Gibbs free energies.25 The second method is indirect, and is based on the thermodynamic cycle for proton exchange reactions, commonly employed for the calculations of pKa values.12 With the indirect method, the gas-phase free energies of reactants and products are corrected by solvation energies and energies due to the geometry relaxation in the solvent. These corrections are straightforwardly acquired from energy differences between the gaseous and solvated states, since COSMO implementation in ADF reports the energies relative to the gas phase fragments.17Acknowledgements
This work was supported by the Serbian Ministry of Education, Science and Technological Development (Project No. 172027) and by the Serbian Academy of Sciences and Arts (Project No. F193). The authors gratefully acknowledge the support of the humanitarian foundation “Hrast” and the support of Dr Ivan Trifunovic. The authors acknowledge the support of the FP7 RegPot project FCUB ERA GA No. 256716. The EC does not share responsibility for the content of the article.Notes and references
- For recent reviews, see: (a) R. K. Haynes, K.-W. Cheu, D. N'Da, P. Coghi and D. Monti, Infect. Disord.: Drug Targets, 2013, 13, 217 CrossRef CAS; (b) D. M. Opsenica and B. A. Solaja, Maced. J. Chem. Chem. Eng., 2012, 31, 137 CAS; (c) R. D. Slack, A. M. Jacobine and G. H. Posner, Med. Chem. Commun., 2012, 3, 281 RSC; (d) L. Tilley, S. A. Charmen and J. L. Vennerstrom, Semisynthetic artemisinin and synthetic antimalarials, in RSC Drug Discovery Series No. 14, Neglected Deseases and Drug Discovery, ed. M. Palmer and T. N. C. Wells, Royal Society of Chemistry, 2012 Search PubMed.
- G. Chianese, F. Scala, B. Calcinai, C. Cerrano, H. A. Dien, M. Kaiser, D. Tasdemir and O. Taglialatela-Scafati, Mar. Drugs, 2013, 11, 3297 CrossRef PubMed.
- A review article on anticancer activity of artemisinin and congeners: A. K. Das, Ann. Med. Health Sci. Res., 2015, 5, 93 CrossRef CAS PubMed.
- A review article on naturally occuring, bioactive peroxides: M. Jung, H. Kim, K. Lee and M. Park, Mini-Rev. Med. Chem., 2003, 3, 159 CrossRef CAS PubMed.
- (a) For the most recent, comprehensive review article on synthesis of endoperoxides, see: A. O. Terent'ev, D. A. Borisov, V. A. Vil and V. M. Dembitsky, Beilstein J. Org. Chem., 2014, 10, 34 CrossRef PubMed, and references therein; (b) E. E. Korshin and M. Bachi, Synthesis of Cyclic Peroxides, in Chemistry of Peroxides, ed. Z. Rappoport, John Wiley & Sons, 2006, Chichester, p. 189, vol. 2 Search PubMed.
- J. E. Baldwin, D. H. R. Barton, D. J. Faulkner and J. F. Templeton, J. Chem. Soc., 1962, 4743 CAS.
- 1,2-Dioxane-3,5-diol structural unit (17, Fig. 1) has been found in a degradation product 18 of artemisinin [quinghaosu, 19; W. S. Zhou, L. Zhang, Z.-C. Fan and X.-X. Xu, Tetrahedron, 1986, 42, 4437 CrossRef CAS ] and has been studied as an intermediate in an alternative synthesis of this well known antimalarial drug [ G. R. Clark, M. M. Nikaido, C. K. Fair and J. Lin, J. Org. Chem., 1985, 50, 1994 CrossRef ; for total syntheses of artemisinin, see: J. S. Yadav, B. Thirupathaiah and P. Srihari, Tetrahedron, 2010, 66, 2005 CrossRef , and references therein]. Interestingly, the same structural unit was found in steenkrotin B (20) – a natural product recently isolated from Croton steencampianus. A. M. Adelekan, E. A. Prozesky, A. A. Hussein, L. D. Urefia, P. H. van Rooyen, D. C. Liles, J. J. M. Meyer and B. Rodriguez, J. Nat. Prod., 2008, 71, 1919.
. - H. C. Brown, J. H. Brewster and H. Schechter, J. Am. Chem. Soc., 1954, 76, 467 CrossRef CAS.
- (a) For a review article on the α-hydrooxylation of enolates and silyl enol ethers, including reactions of enolates with oxygen, see: B. C. Chen, P. Zhou, F. A. Davis and E. Ciganek, Org. React, J. Wiley & Sons, Hoboken, 2003, p. 1, vol. 62 Search PubMed; (b) for a recent study on the mechanism of base-catalyzed autooxidation of steroidal ketones, see: M. Li, B. Chen, S. Monteiro and A. M. Rustum, Tetrahedron Lett., 2009, 50, 4575 CrossRef CAS.
- H. R. Gersmann and A. F. Bickel, J. Chem. Soc. B, 1968, 2978 Search PubMed.
- M. Swart, E. Rösler and F. M. Bickelhaupt, Eur. J. Inorg. Chem., 2007, 2007, 3646 CrossRef.
- C. C. R. Sutton, G. V Franks and G. da Silva, J. Phys. Chem. B, 2012, 116, 11999 CrossRef CAS PubMed.
- For details, see Computational Details section in the Experimental part.
- For aryl ketones, DFT calculations are known to overestimate the contribution of conjugation with aromatic core to the enolate stability.
- (a) G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS; (b) C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391 Search PubMed; (c) ADF: Density Functional Theory (DFT) software for chemists version 2013.01, http://www.scm.com/, 2013, accessed Oct. 2015 Search PubMed.
- (a) A. Klamt and G. Schoermann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 RSC; (b) A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- J. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1988, 88, 2547 CrossRef CAS; (b) M. Franchini, P. H. T. Philipsen and L. Visscher, J. Comput. Chem., 2013, 34, 1819 CrossRef CAS PubMed.
- (a) A. Bérces, R. M. Dickson, L. Fan, H. Jacobsen, D. Swerhone and T. Ziegler, Comput. Phys. Commun., 1997, 100, 247 CrossRef; (b) H. Jacobsen, A. Bérces, D. P. Swerhone and T. Ziegler, Comput. Phys. Commun., 1997, 100, 263 CrossRef CAS; (c) S. K. Wolff, Int. J. Quantum Chem., 2005, 104, 645 CrossRef CAS.
- (a) J. Ho, Phys. Chem. Chem. Phys., 2015, 17, 2859 RSC; (b) J. H. Jensen, Phys. Chem. Chem. Phys., 2015, 17, 12441 RSC; (c) A. Klamt, B. Mennucci, J. Tomasi, V. Barone, C. Curutchet, M. Orozco and F. J. Luque, Acc. Chem. Res., 2009, 42, 489 CrossRef CAS PubMed ; discussion 493.
- M. Swart, E. Rösler and F. M. Bickelhaupt, Eur. J. Inorg. Chem., 2007, 2007, 3646 CrossRef.

Footnotes |
| † Dedicated to the memory of our colleague and friend Dr Suren Husinec (1953–2015). |
| ‡ Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all compounds, CIF files for compounds 2, 6c, 6e and 6f are deposited. CCDC 1411164–1411167. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra13476e |
| This journal is © The Royal Society of Chemistry 2015 |