DOI:
10.1039/C5RA13416A
(Paper)
RSC Adv., 2015,
5, 77460-77468
Design, synthesis and electronic properties of push–pull–push type dye†
Received
9th July 2015
, Accepted 7th September 2015
First published on 7th September 2015
Abstract
A Sonogashira cross-coupling protocol was employed for the construction of a push–pull–push type dye. An ethynyl π-spacer extends the effective π-conjugation length between push and pull units without altering the planarity of the electron donor/acceptor pair. The variation of the strength of the alkyne π-spacer electron push (or donor) units of these dyes has a strong effect on the shifting of both absorption and emission maxima and thereby on the Stokes shift. The dyes were solvatochromic and their solvatochromicity was highly dependent on the electron push unit. Strong red shifted emissions were likely to arise due to the internal charge transfer (ICT) from the electron push unit to the electron pull unit. Calculated energy values of HOMO → LUMO transitions are in good accordance with experimental observations. Alkyne conjugated electron push units (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Ar; Ar = Ph, Ph–OMe, Ph–NMe2) are more effective to increase the EHOMO levels. Overall, experimental and theoretical results of the push–pull–push dyes indicate that they can be used as promising conjugated materials with predictable electronic properties for optoelectronic devices.
C–Ar; Ar = Ph, Ph–OMe, Ph–NMe2) are more effective to increase the EHOMO levels. Overall, experimental and theoretical results of the push–pull–push dyes indicate that they can be used as promising conjugated materials with predictable electronic properties for optoelectronic devices.
Introduction
π-Conjugated donor–acceptor type molecules have been widely used in material science as an efficient organic electronics material for applications in organic light-emitting diodes (OLEDs),1–5 organic field effect transistors (OFETs),6–13 organic photovoltaic devices (OPVs)14–29 and so forth. Ethynyl (–CC–) and vinyl (–HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) π-conjugates are structurally very similar and could potentially reduce the band gap energy.30–35 However, vinyl π-conjugated systems show better charge delocalization and smaller band gap energy over the ethynyl π-conjugation.36 Indeed, an ethynyl π-spacer is sterically less hindered and more accommodating than a vinyl π-spacer.37 It is also noteworthy that the steric crowd between face-to-face groups of vinyl and aromatic units result a non-planar conformation, which reduces the charge delocalization efficiency.38,39 Additionally, the incorporation of an ethynyl π-spacer is a straightforward approach and easier than a vinyl π-spacer in synthetic point of view.40,41 Thus, design and synthesis of novel ethynyl π-conjugated donor–acceptor type molecules having high performance material properties remain a promising area of optoelectronic research. The push–pull–push type dye structure, where the electron donating units push the negative charge density and electron accepting units pull the negative charge density towards itself. Both the push and pull units are connected by π-conjugated alkyne (–CC–) spacer unit which enables the intramolecular charge transfer (ICT) from electron rich (push) to electron deficient (pull) units, and thereby facilitates red shifted absorbance with high molar extinction coefficient.42–51 The electron deficient unit tunes the energy levels of frontier molecular orbitals and at the same time the π-conjugated alkyne spacer connected electron rich unit also involves to monitor the energy levels.17 According to the donor capability of the donating unit, we can monitor the energy level of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) and can obtain different physical properties (Fig. 1).17,52–59
CH–) π-conjugates are structurally very similar and could potentially reduce the band gap energy.30–35 However, vinyl π-conjugated systems show better charge delocalization and smaller band gap energy over the ethynyl π-conjugation.36 Indeed, an ethynyl π-spacer is sterically less hindered and more accommodating than a vinyl π-spacer.37 It is also noteworthy that the steric crowd between face-to-face groups of vinyl and aromatic units result a non-planar conformation, which reduces the charge delocalization efficiency.38,39 Additionally, the incorporation of an ethynyl π-spacer is a straightforward approach and easier than a vinyl π-spacer in synthetic point of view.40,41 Thus, design and synthesis of novel ethynyl π-conjugated donor–acceptor type molecules having high performance material properties remain a promising area of optoelectronic research. The push–pull–push type dye structure, where the electron donating units push the negative charge density and electron accepting units pull the negative charge density towards itself. Both the push and pull units are connected by π-conjugated alkyne (–CC–) spacer unit which enables the intramolecular charge transfer (ICT) from electron rich (push) to electron deficient (pull) units, and thereby facilitates red shifted absorbance with high molar extinction coefficient.42–51 The electron deficient unit tunes the energy levels of frontier molecular orbitals and at the same time the π-conjugated alkyne spacer connected electron rich unit also involves to monitor the energy levels.17 According to the donor capability of the donating unit, we can monitor the energy level of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) and can obtain different physical properties (Fig. 1).17,52–59
 |
| | Fig. 1 Chemical structures of push–pull–push dye. | |
In our study, we demonstrate the incorporation of an alkyne π-spacer between the donor (push) and acceptor (pull) units. The alkyne π-spacer was introduced to extend the effective conjugation, and improve the photophysical properties.31,60,61 In addition, the planer structure of alkyne facilitates efficient charge transport which can improve the optoelectronic properties. We chose the readily synthesizable thieno-[3,4-c]pyrrole-4,6-dione (TPD) as an electron pull unit which is symmetric, rigidly fused planer structure having strong electron withdrawing property.62–72 This unit provides low energy HOMO level, low band gap, and is convenient for the introduction of variety of alkyl side chains into the pyrrole ring to improve the solubility in organic solvents. Varieties of ethynyl donar groups (Ar–CC–) are commercially available or easily synthesizable according to the requirement. Sonogashira cross-coupling gives the facile synthetic advantage of structural variation in the electron push unit of the push–pull–push dye. Overall, the dyes showed tunable electronic properties. Herein, we investigated the effect on variation of the strength of alkyne π-spacer electron donor units of the push–pull–push dye using a combination of experimental and theoretical studies.
Results and discussion
Synthesis
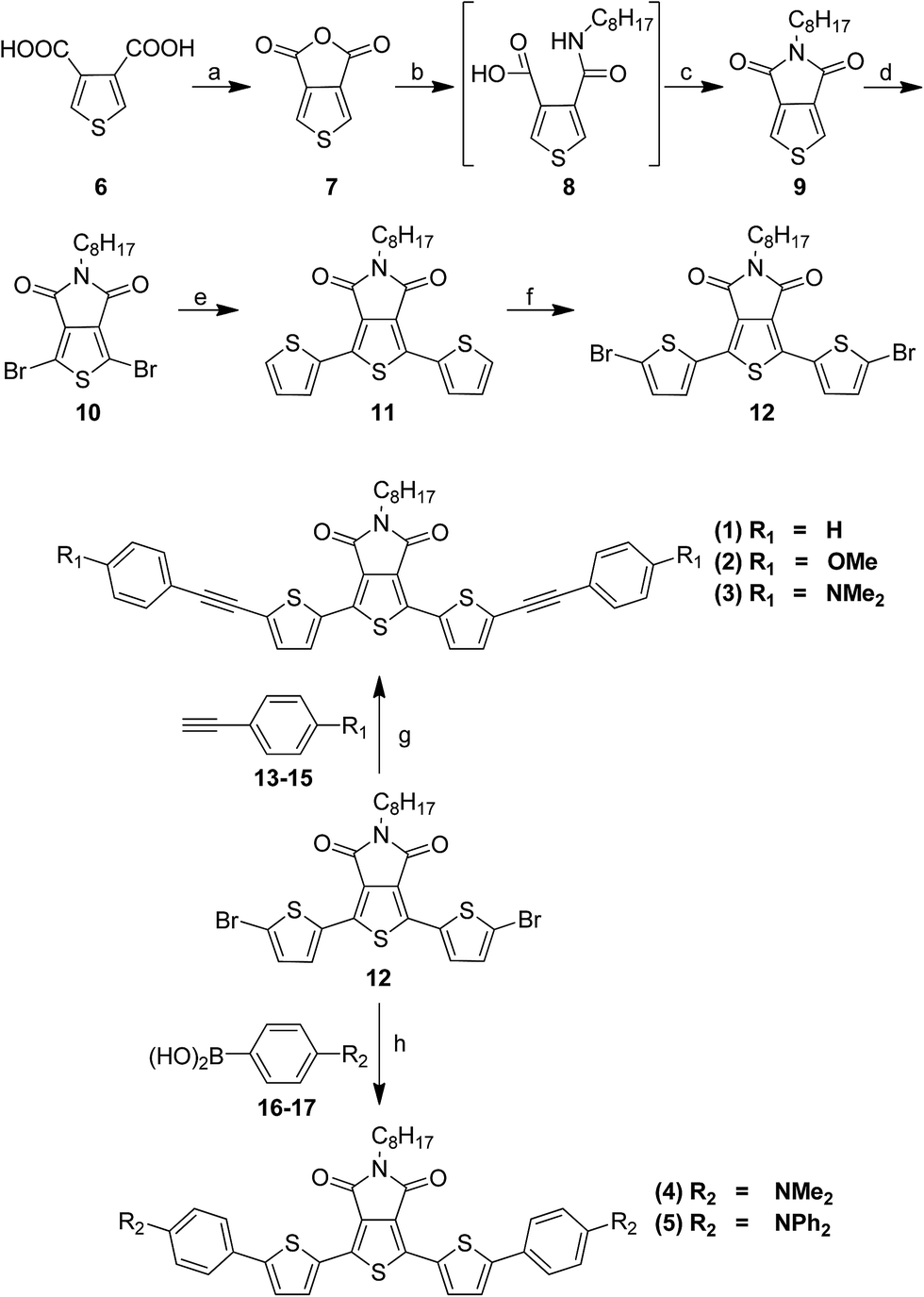
The push–pull–push dyes were synthesized by cross coupling reaction between the donor (push) and acceptor (pull) units and are shown in Scheme 1. Sonogashira coupling40,41 was used to synthesize alkyne π-spacer conjugated dyes and Suzuki coupling73 was used to attach the donor and acceptor units directly via C(sp2)–C(sp2) coupling without alkyne π-spacer. The TPD acceptor unit was synthesized from thiophene-3,4-dicarboxylic acid.62–64 Thus, the refluxing of thiophene-3,4-dicarboxylic acid (6) in acetic anhydride gave thiophene-3,4-dicarboxylic anhydride (7), which when treated with n-octylamine followed by stirring with thionyl chloride afforded the TPD unit, 5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (9). Compound 9 was brominated to give dibromo product (10), which was further reacted with 2-(tributylstannyl)thiophene to yield 5-octyl-1,3-di(thiophen-2-yl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (11). Bromination of 11 using N-bromosuccinimide and prolonged reaction time resulted in symmetric dibromo compound 12. Finally, the dyes 1–3 were synthesized via C(sp2)–C(sp3) Sonogashira coupling between 12 and terminal alkynes 13–15. Whereas the dyes 4–5 were synthesized via C(sp2)–C(sp2) Suzuki coupling between 12 and boronic acids 16 and 17. The intermediate and final products were characterized by NMR and ESI MS (see ESI†). The very pure and recrystallized form of the dyes (1–5) were used to study the photophysical and optoelectronic properties.
 |
| | Scheme 1 Synthesis of push–pull–push dye 1–5. Reagents and conditions: (a) (CH3CO)2O, heat 140 °C, 12 h; (b) C8H17NH2, toluene, reflux, 24 h; (c) SOCl2, 50 °C, 8 h; (d) N-bromosuccinimide (NBS), H2SO4, CF3COOH, 10 h; (e) 2-(tributylstannyl)thiophene, Pd2(dba)2, tri(O-tolyl)phosphine, toluene, reflux, 18 h; (f) NBS, CHCl3, CH3COOH, 0 °C to rt, 30 h; (g) Pd(PPh3)4, CuI, N,N-diisopropylethylamine (DIPEA), DMF, 80 °C, 24 h; (h) Pd(PPh3)2Cl2, K3PO4, DMF, 80 °C, 24 h. | |
Photophysical properties
First, we investigated the absorption photophysical properties of the dyes (1–5) in different solvents of varying dielectric constants. We observed 10 to 20 nm bathochromic shift of all the dyes upon changing the solvent polarity from nonpolar hexane to polar solvent dimethyl sulfoxide (DMSO). However, in polar protic solvent MeOH, we observed hypsochromic shift, and this is because of protic solvent–solute interaction between the dye and the solvent.74,75
Inconsistency behaviour in the absorption maxima was shown in some of the intermediate polar solvents. Donating groups have a strong effect on monitoring the absorptive properties of the dyes 1–3. The dye 1 containing phenyl group as the electron push unit shows average absorption maxima at around 440 nm and lies between 433–444 nm; whereas, dye 3 containing 4-N,N-dimethylaniline group as electron push unit showed absorption maxima at around 470 nm and lies between 465–485 nm (Fig. 2a and e). The intermediate electron push unit 4-methoxyphenyl shows absorption maxima at around 450 nm and lies between 446–456 nm (Fig. 2c). Thus, we observed ∼10 nm bathochromic shift from the dye 1 to 2, by changing the electron push unit phenyl (–Ph–H) to 4-methoxyphenyl (–Ph–OMe) group and ∼30 nm bathochromic shift from the dye 1 to 3, by changing phenyl (–Ph–H) to more efficient electron push unit 4-N,N-dimethylaniline (–Ph–NMe2) group. The dyes 4 and 5 absorb between 468–488 nm. However, the molar extinction coefficient of the dye 5 is more and almost doubled compared to the dye 4 and this is because –NPh2 group in dye 5 provides larger conjugated system than –NMe2 group provides in dye 4 (Fig. 3a and c).
 |
| | Fig. 2 UV-visible (a, c and e) and fluorescence (b, d and f) spectra of the dyes 1, 2 and 3 respectively in different solvents (10 μM, RT, λex = λUV max of each solvents). | |
 |
| | Fig. 3 UV-visible (a and c) and fluorescence (b and d) spectra of the dyes 4 and 5 respectively in different solvents (10 μM, RT, λex = λUV max of each solvents). | |
Next, we evaluated the fluorescence emission behaviour of the dyes (1–5) in different solvents moving from nonpolar hexane to polar MeOH solvents (Fig. 2b, d, f, 3b and d). We observed very interesting solvent dependent emission properties upon changing the electron push unit. The emission properties of the dyes were more effective on solvent polarity than the absorption properties. All the dyes showed structured band in low polar or medium polar solvents and structureless broad emission band in high polar solvents. Interestingly, for all the cases we observed strong red shifted fluorescence emission with decrease in fluorescence intensity as the solvent polarity changes from nonpolar to polar. These results clearly indicate that the emission bands were arise due to the internal charge transfer (ICT) from electron push unit to electron pull unit. The large Stokes shift was observed with increasing solvent polarity of all the dyes and it was also highly affected by the electron push unit and the π-conjugation (Table 1, and see ESI†). The electron donor units used in this study follow the order –Ph–H < –Ph–OMe < –Ph–NPh2 ≤ –Ph–NMe2. With increasing solvent polarity a red shift of about 20 nm (λmax,hexane = 487 nm and λmax,DMSO = 507 nm) for the dye 1 (containing phenyl (–Ph–H) as electron push unit) was observed. Replacement of the phenyl (–Ph–H) group in dye 1 with a strong electron donating 4-N,N-dimethylaniline (–Ph–NMe2) group in dye 3 leads to a strong red shift of about 75 nm (λmax,hexane = 520 nm and λmax,EtOAc = 595 nm). The moderate electron donating group 4-methoxyphenyl (–Ph–OMe) containing dye 2 showed 40 nm red shift from hexane (λmax,hexane = 500 nm) to dichlorobenzene (λmax,dichlorobenzene = 540 nm) (see ESI†). As –Ph–NMe2 group is the strongest electron donating group among the four, so best charge delocalization through the alkyne π-spacer occurs in dye 3 and it shows highest absorption and emission maxima and also lowest band gap energy. Thus, from the above observations it is clear that the electron push units of these push–pull–push dyes have a strong effect towards the shifting of both absorption and emission maxima and thereby on the Stokes shift. The lack of alkyne π-spacer conjugation reduces the red shift to 45 nm (λmax,hexane = 533 nm and λmax,EtOAc = 578 nm) in dye 4 although it contains the strong electron donating 4-N,N-dimethylaniline (–Ph–NMe2) group. Dye 5 containing the electron push unit 4-N,N-diphenylaniline (–Ph–NPh2) group showed 85 nm (λmax,hexane = 530 nm and λmax,DMSO = 615 nm) red shift although it does not contain the alkyne π-spacer conjugation. The large conjugated system due to the –NPh2 group compensates the lack of alkyne π-spacer conjugation and leads to a strong red shifted emission. Both the fluorescence intensity and the quantum yield were decreased with increasing solvent polarity for all the dyes (1–5). All the above observations revealed that the dyes were solvatochromic and their solvatochromicity was highly dependent on the electron push unit and the π-conjugation. The photophysical properties of the dyes (1–5) in CHCl3 solutions are summarized in Table 1.
Table 1 Photophysical propertiesa of the dye 1–5
| Dye |
λmax(Abs) (nm) |
εmax M−1 cm−1 |
λmax(Em) (nm) |
Stokes shift (cm−1) |
Φfb |
| Photophysical data in CHCl3 solvent. Quantum yield using fluorescein in 0.1 N NaOH as a standard (Φf = 0.92), λex = 440, 453, 480 nm. |
| 1 |
440 |
45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
501 |
2767 |
0.119 |
| 2 |
453 |
37100 |
529 |
3171 |
0.142 |
| 3 |
485 |
30600 |
581 |
3406 |
0.096 |
| 4 |
477 |
18000 |
566 |
3296 |
0.096 |
| 5 |
482 |
62500 |
572 |
3264 |
0.071 |
The solvatochromicity of the dyes (1–5) was reflected on the colour of the fluorescence images, which changed in different solvents under a 254 nm wavelength transilluminator (see ESI†). The influence of electron donating units on the shifting of emission maxima of these push–pull–push dyes was also nicely observed by the colour change in chloroform solvent under a 254 nm wavelength transilluminator (Fig. 4). The colour varies from cyan to green to yellow to orange region accordingly with the electron donating capacity (–Ph–H < –Ph–OMe < –Ph–NPh2 ≤ –Ph–NMe2) of the electron push unit and the extra π-conjugation effect.
 |
| | Fig. 4 Normalized (a) UV-visible and (b) fluorescence spectra the dyes 1–5 in CHCl3 solvents (10 μM, RT, λex = λUV max). (c) Colours: colours in CHCl3 solvents after irradiation of 1–5 in UV light (λ = 254 nm) under a UV-transilluminator. | |
After solution state photophysical study, we investigated the solid state photophysical properties of the dyes (1–5) as film. To measure thin films, dyes were spin coated on pre-cleaned quartz substrates using chloroform solutions. To ensure the smooth films, spin coating time and speed were fixed at 60 s and 1000 rpm. It is noteworthy that both the solution and solid state results follow similar trends. The UV-visible absorption and fluorescence spectra of the dyes (1–5) in the film state are shown in Fig. 5. The dyes 1–5 exhibit absorption peak centered at 456, 456, 494, 480, and 485 nm respectively. Thus, all the synthesized push–pull–push dyes (1–5) absorbs visible light. The fluorescence emission maxima of the dyes 1–5 centered at 583, 610, 652, 623, and 638 nm respectively. Dye 3 showed highest absorption and emission maxima among all the dyes 1–5, which can be attributed to increase in strength of the electron donating group and π-conjugation. Thus, the effect of electron donating groups and the of alkyne π-spacer conjugation also reflected on the photophysical properties in film state.
 |
| | Fig. 5 Normalized (a) UV-visible and (b) fluorescence spectra of the dyes 1–5 as film, spin coated on pre-cleaned quartz substrates using chloroform solutions. | |
Computational studies
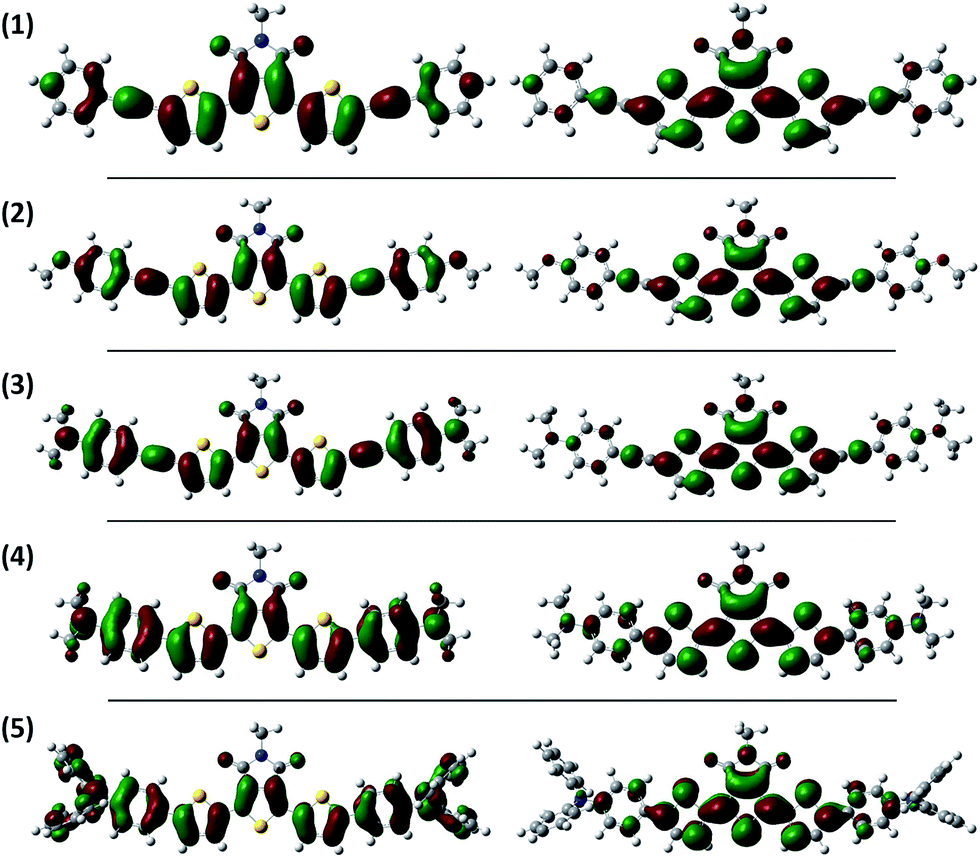
Highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) of push–pull–push dye (1–5) have been calculated using Gaussian 03 program package.76 First the geometries were optimized using the B3LYP hybrid density functional and 3-21G* as the basis set. Next, the optimized geometries were used for TD-SCF calculation using the same B3LYP hybrid density functional and 3-21G* as the basis set. For all the dyes, HOMO resides mainly on the electron push units and/or alkyne π-spacer including thiophene units. In contrast, LUMO resides mainly on the TPD moiety and delocalized nearby two thiophene units attached with TPD moiety. The electron density diagrams of HOMO and LUMO are shown in Fig. 6. The calculated allowed transitions are listed in Table 2. The lowest energy associated with strong absorption band arises from S0 → S1 transition which is attributed to the electronic transition from HOMO → LUMO.
 |
| | Fig. 6 HOMOs (left) and LUMOs (right) of push–pull–push dye (1–5) (calculated using Gaussian 03; Opt + Freq: B3LYP/3-21G*//TD-SCF: B3LYP/3-21G*). | |
Table 2 Calculated lowest energy S0 → S1 transition
| Dye |
Electronic transition |
Wavelength (nm) |
Oscillator strengths |
Major contributions HOMO → LUMO |
| 1 |
S0 → S1 |
485 |
1.8147 |
83.9% |
| 2 |
S0 → S1 |
505 |
1.9778 |
85.0% |
| 3 |
S0 → S1 |
546 |
2.0238 |
86.9% |
| 4 |
S0 → S1 |
510 |
1.5026 |
86.5% |
| 5 |
S0 → S1 |
528 |
1.6926 |
88.8% |
Thus, all these low energy strong absorptions mostly (84% to 89%) originated from HOMO → LUMO correspond to π → π* transitions which arise due to intramolecular charge transfer (ICT) from electron push units to electron pull TPD unit through the π-conjugation. The calculated energy values for S0 → S1 transition of the dye 1–5 were in good agreement with the experimental absorption energy values (Tables 1 and 2).
Cyclic voltammetry studies
To evaluate the energy parameters of the dye 1–5, cyclic voltammetry (CV) measurements were carried out. Ferrocene/ferrocenium (Fc/Fc+) redox couple was used as an internal reference. The HOMO and LUMO energy levels were calculated from the onset of the first oxidation and reduction potentials using the equations EHOMO (eV) = −[Eonsetox − E1/2 (Fc/Fc+) + 4.8] and ELUMO (eV) = −[Eonsetred − E1/2 (Fc/Fc+) + 4.8], where E1/2 (Fc/Fc+) was the cell correction. It showed two peaks at 0.389 and 0.485 V hence the E1/2 (Fc/Fc+) was equal to 0.437 V, which was used in equation to calculate the EHOMO and ELUMO. The energy parameters of the dye (1–5) are summarized in Table 3 (see ESI† for the cyclic voltammogram). Thus, the summarized EHOMO and ELUMO values show that the alkyne conjugated electron push unit (–CC–Ar; Ar = Ph, Ph–OMe, Ph–NMe2) is more effective to increase the EHOMO levels. However, the ELUMO levels are not significantly affected by the electron push unit. It is noteworthy that band gap energy decreases upon introducing electron donor units into 11. The decrease in ELUMO and increase in EHOMO levels of the dyes (1–5) upon introducing electron donor units into 11 can be explained in terms of inductive effect and resonance effect.17 TPD is a strong electron withdrawing unit and shows strong inductive effect. The conjugated electron donor units show resonance effect and able to delocalize the charge density and or π-system with neighbouring ethynyl π-spacer and or thiophene unit. Thus, both the inductive effect and resonance effect increases the effective delocalization which strongly affect the EHOMO and ELUMO levels to reduce the band gap energy of the dyes (1–5).
Table 3 Energy parameters of the dye 1–5
| Dye |
Eox (V) |
Ered (V) |
EHOMO (eV) |
ELUMO (eV) |
Eox–red (eV) |
| 1 |
1.28 |
−0.754 |
−5.643 |
−3.609 |
2.034 |
| 2 |
1.11 |
−0.689 |
−5.473 |
−3.674 |
1.799 |
| 3 |
0.826 |
−0.76 |
−5.189 |
−3.603 |
1.586 |
| 4 |
0.856 |
−0.73 |
−5.219 |
−3.633 |
1.586 |
| 5 |
0.566 |
−0.853 |
−4.929 |
−3.51 |
1.419 |
Overall, the electron donor units of these push–pull–push dye carefully balance both the EHOMO and ELUMO levels, which make them promising optoelectronics material.
Conclusions
In summary, we have synthesized push–pull–push type dyes and investigated the electronic properties using a combination of both experimental and theoretical studies. Ethynyl π-spacer improved the electronic properties of the dye by extending the effective π-conjugation between electron donor/acceptor units. All the dyes showed highly solvent dependent red shifted emission and large Stokes shift. The solvatochromism of the dyes significantly depends on the strength of the electron donor unit. The red shifted emissions were assigned as internal charge transfer (ICT) between electron donor/acceptor pair. The influence of electron donating units on the shifting of emission maxima of these push–pull–push dyes were reflected on the colour change in chloroform solvent under a 254 nm wavelength transilluminator. Strong electron donating 4-N,N-dimethylaniline (–Ph–NMe2) group in dye 3 leads to a strong red shift of about 75 nm (λmax,hexane = 520 nm and λmax,EtOAc = 595 nm). Calculated energy values of HOMO → LUMO transitions are in good agreement with experimental observations. The electron push units (–CC–Ar; Ar = Ph, Ph–OMe, Ph–NMe2) are more effective to increase the EHOMO levels. However, the ELUMO levels are not significantly affected by the electron push unit. Overall, findings indicate that these push–pull–push dyes can be used as a promising conjugated materials with predictable electronic properties for optoelectronic devices.
Experimental section
General
All the reagents and solvents were purchased from commercial suppliers and were used without further purification. The air- and/or moisture-sensitive reactions were carried out under nitrogen atmosphere. All the reactions were monitored with analytical TLC silica gel 60 F254. The products were purified by silica gel (230–400 mesh size) column chromatography. 1H and 13C NMR spectra were recorded under 400 MHz and 100 MHz Bruker Advance NMR spectrometer respectively using CDCl3 as the solvent. All the UV-visible spectra of the compounds (10 μM) were measured in different solvents using a UV-visible spectrophotometer at room temp with a cell of 1 cm path length. All the sample solutions prepared for UV-visible experiments were further used for fluorescence experiments immediately after UV-visible experiments. The fluorescence spectra were obtained using a fluorescence spectrophotometer at room temp using 1 cm path length cell. The fluorescence quantum yields (Φf) were determined using fluorescein as a reference with the known Φf (0.92) in 0.1 molar solution in sodium hydroxide. The electrochemical cyclic voltammetry (CV) was conducted on a PowerLab/AD instrument model system with glassy carbon disk, Pt wire, and Ag/Ag+ electrode as the working electrode, counter electrode, and reference electrode, respectively in a 0.1 M tetrabutylammonium hexafluorophosphate (n-Bu4NPF6)–anhydrous acetonitrile solution at a potential scan rate of 50 mV s−1. Material film was drop cast onto the glassy carbon working electrode from a 2.0 mg mL−1 chloroform solution and dried. The electrochemical onsets were determined at the position where the current starts to differ from the baseline. The potential of Ag/AgCl reference electrode was internally calibrated by using the ferrocene/ferrocenium redox couple (Fc/Fc+). The HOMO and LUMO energy levels were calculated from the onset of the first oxidation and reduction potentials using the equations EHOMO (eV) = −[Eonsetox − E1/2 (Fc/Fc+) + 4.8] and ELUMO (eV) = −[Eonsetred − E1/2 (Fc/Fc+) + 4.8], where E1/2 (Fc/Fc+) was the cell correction. Gaussian 03 program package was used for the computational study. The geometries were optimized using the B3LYP hybrid density functional and 3-21G* as the basis set. Next, the optimized geometries were used for TD-SCF calculation using the same B3LYP hybrid density functional and 3-21G* as the basis set. For all the cases imaginary frequency values after opt-freq (optimization followed by frequency) calculation were zero.
Thieno[3,4-c]furan-1,3-dione (7). A solution of 4 g (23.242 mmol) thiophene-3,4-dicarboxylic acid (6) in 150 mL acetic anhydride was refluxed for 12 h. After the completion of reaction, the solution was evaporated under reduced pressure and the solid residue was recrystallized from hot toluene, 3.402 g, 95% yield. 1H NMR (400 MHz, CDCl3) δ 8.10 (2H, s); 13C NMR (100 MHz, CDCl3) δ 129.5, 135.3, 156.5; HRMS calcd for C6H2O3S 153.9725, found 153.9711.
5-Octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (9). To a solution of 3.35 g (21.739 mmol) thieno[3,4-c]furan-1,3-dione (7) in toluene, 7.2 mL n-octylamine was added and refluxed for 24 h. The reaction mixture was cooled down and the solvent was evaporated under reduced pressure. The crude solid was dissolved in 200 mL thionyl chloride (SOCl2) and stirred for 8 h under 50 °C. The volatile solvent was removed and the crude product was purified by silica gel (230–400 mesh size) column chromatography using hexane–dichloromethane as eluent to afford the white solid product, 3.75 g, 65% yield. 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, t, J = 5.2 Hz), 1.25–1.31 (10H, m), 1.61–1.67 (2H, m), 3.60 (2H, t, J = 7.2 Hz), 7.79 (2H, s); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.7, 27.0, 28.6, 29.2, 31.9, 38.6, 125.5, 136.8, 162.7; HRMS calcd for C14H19NO2S 265.1136, found 265.1125.
1,3-Dibromo-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (10). To a solution of 3.7 g (13.946 mmol) 5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (9) in a mixture of sulfuric acid (10 mL) and trifluoroacetic acid (30 mL), 5.46 g (30.681 mmol) N-bromosuccinimide was added in eight portions. The reaction mixture was stirred at room temperature for 10 h. The reddish brown solution was diluted with water (120 mL) and the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous magnesium sulfate followed by concentrated under reduced pressure. The crude product was purified by silica gel (230–400 mesh size) column chromatography using hexane–dichloromethane as eluent to afford the white crystalline solid product, 4.72 g, 80% yield. 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, t, J = 6.4 Hz), 1.26–1.30 (10H, m), 1.60–1.64 (2H, m), 3.58 (2H, t, J = 7.6 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.6, 26.8, 28.2, 29.1, 31.7, 38.7, 112.7, 134.8, 160.1; HRMS calcd for C14H17Br2NO2S 420.9347, found 420.9338.
5-Octyl-1,3-di(thiophen-2-yl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (11). To a solution of 4.5 g (10.635 mmol) 1,3-dibromo-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (10) in toluene, 2-(tributylstannyl)thiophene (10.317 g, 8.78 mL, 27.652 mmol), tris(dibenzylideneacetone)dipalladium(0) (146.5 mg, 0.16 mmol, 1.5 mol%) and tri(O-tolyl)phosphine (194.1 mg, 0.638 mmol, 6 mol%) were added under nitrogen atmosphere. The reaction mixture was reflux for 18 h. After completion of the reaction, the mixture was extracted with dichloromethane. The organic layer was dried over anhydrous magnesium sulfate followed by concentrated under reduced pressure. The crude product was purified by silica gel (230–400 mesh size) column chromatography using hexane–dichloromethane as eluent to afford the yellow powdered product, 3.29 g, 72% yield. 1H NMR (400 MHz, CDCl3) δ 0.86 (3H, t, J = 6.8 Hz), 1.26–1.34 (10H, m), 1.63–1.71 (2H, m), 3.66 (2H, t, J = 7.2 Hz), 7.12–7.14 (2H, m), 7.44 (2H, d, J = 5.2 Hz), 8.01 (2H, d, J = 3.6 Hz); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.7, 27.1, 28.6, 29.3, 31.9, 38.7, 128.5, 128.7, 130.0, 132.5, 136.6, 162.7; HRMS calcd for C22H23NO2S3 429.0891, found 429.0885.
1,3-Bis(5-bromothiophen-2-yl)-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (12). To a solution of 3.2 g (7.448 mmol) 5-octyl-1,3-di(thiophen-2-yl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (11) in 1:1 mixture of CH3COOH–CHCl3 under ice bath, 3.314 g (18.62 mmol) N-bromosuccinimide was added in eight portions. The reaction mixture was allowed to come at RT and stirred for 30 h. The solvent was evaporated and the residue was purified by silica gel (230–400 mesh size) column chromatography using hexane–dichloromethane as eluent to afford the yellow powdered product, 3.5 g, 80% yield. 1H NMR (400 MHz, CDCl3) δ 0.86 (3H, t, J = 7.2 Hz), 1.26–1.31 (10H, m), 1.62–1.67 (2H, m), 3.61 (2H, t, J = 7.2 Hz), 7.05 (2H, d, J = 4.0 Hz), 7.63 (2H, d, J = 4.0 Hz); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.8, 27.1, 28.6, 29.3, 31.9, 38.8, 116.9, 128.7, 129.9, 131.3, 133.9, 135.2, 162.4; HRMS calcd for C22H21Br2NO2S3 584.9101, found 584.9113.
General procedure for the Sonogashira coupling
To a solution of aryl halide 12 (1,3-bis(5-bromothiophen-2-yl)-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione) in 1:1 DMF–Et3N under nitrogen atmosphere, Pd(PPh3)4 (6 mol%), CuI (3 mol%) and terminal alkyne (13–15, 2.6 equiv.) were added. The reaction mixture was stirred for 24 h at 80 °C and monitored by TLC. After the completion of the reaction, the solvent was evaporated and the resulting residue was purified by silica gel (230–400 mesh size) column chromatography using hexane–dichloromethane as eluent to afford the product. The products were isolated with very good yield and characterized by NMR, and mass spectrometry.
General procedure for the Suzuki coupling
To a solution of aryl halide 12 (1,3-bis(5-bromothiophen-2-yl)-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione) in DMF under nitrogen atmosphere, PdCl2(PPh3)2 (6 mol%), arylboronic acid (16–17, 2.5 equiv.) and potassium phosphate (K3PO4, 6 equiv.) were added. The reaction mixture was stirred for 24 h at 80 °C and monitored by TLC. After the completion of the reaction, the solvent was evaporated and the residue was washed with water and extracted by dichloromethane. The crude product was purified by silica gel (230–400 mesh size) column chromatography using hexane–dichloromethane as eluent. The products were isolated with very good yield and characterized by NMR, and mass spectrometry.
1,3-Bis(5-(phenylethynyl)thiophen-2-yl)-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (1). Using the general procedure, to a solution of aryl halide 12 (100.0 mg, 0.170 mmol) in 1:1 DMF–Et3N under nitrogen atmosphere, Pd(PPh3)4 (11.8 mg, 0.01 mmol, 6 mol%), CuI (1.0 mg, 0.005 mmol, 3 mol%) and phenylacetylene, 13 (45.0 mg, 0.442 mmol) were added. The title compound 1 was isolated as red solid, 86.7 mg, 81% yield. 1H NMR (400 MHz, CDCl3) δ 0.88 (3H, t, J = 6.9 Hz), 1.27–1.33 (10H, m), 1.66–1.68 (2H, m), 3.63 (2H, t, J = 7.5 Hz), 7.18 (2H, d, J = 3.9 Hz), 7.34–7.36 (6H, m), 7.48–7.51 (4H, m), 7.84 (2H, d, J = 3.9 Hz); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.8, 27.1, 28.6, 29.3, 31.9, 38.8, 82.4, 96.5, 122.5, 126.7, 128.6, 128.9, 129.1, 129.9, 131.6, 132.9, 133.4, 135.6, 162.4; HRMS calcd for [C38H31NO2S3 + H]+ 630.1595, found 630.1571.
1,3-Bis(5-((4-methoxyphenyl)ethynyl)thiophen-2-yl)-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (2). Using the general procedure, to a solution of aryl halide 12 (100 mg, 0.170 mmol) in 1:1 DMF–Et3N under nitrogen atmosphere, Pd(PPh3)4 (11.8 mg, 0.01 mmol, 6 mol%), CuI (1.0 mg, 0.005 mmol, 3 mol%) and 4-ethynylanisole 14 (58.4 mg, 0.442 mmol) were added. The title compound 2 was isolated as red solid, 91.5 mg, 78% yield. 1H NMR (400 MHz, CDCl3) δ 0.85 (3H, t, J = 6.6 Hz), 1.23–1.30 (10H, m), 1.64–1.66 (2H, m), 3.63 (2H, t, J = 7.2 Hz), 3.81 (6H, s), 6.86 (4H, d, J = 9.0), 7.17 (2H, d, J = 3.9), 7.43 (4H, d, J = 8.7 Hz), 7.86 (2H, d, J = 3.9 Hz); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.8, 27.1, 28.6, 29.3, 31.9, 38.8, 55.5, 81.2, 96.7, 114.3, 127.3, 128.1, 129.0, 130.0, 132.5, 133.0, 133.2, 135.8, 160.2, 162.6; HRMS calcd for [C40H35NO4S3 + H]+ 690.1806, found 690.1821.
1,3-Bis(5-((4-(dimethylamino)phenyl)ethynyl)thiophen-2-yl)-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (3). Using the general procedure, to a solution of aryl halide 12 (500 mg, 0.851 mmol) in 1:1 DMF–Et3N under nitrogen atmosphere, Pd(PPh3)4 (59.0 mg, 0.051 mmol, 6 mol%), CuI (9.0 mg, 0.026 mmol, 3 mol%) and 4-ethynyl-N,N-dimethylaniline 15 (320.8 mg, 2.212 mmol) were added. The title compound 3 was isolated as red solid, 438.7 mg, 72% yield. 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, t, J = 6.6 Hz), 1.26–1.32 (10H, m), 1.66–1.68 (2H, m), 3.00 (12H, s), 3.66 (2H, t, J = 7.2 Hz), 6.66 (4H, d, J = 9.0 Hz), 7.16 (2H, d, J = 4.2 Hz), 7.39 (4H, d, J = 9.0 Hz), 7.89 (2H, d, J = 3.9 Hz); 13C NMR (100 MHz, CDCl3) δ 14.2, 22.8, 27.1, 28.6, 29.3, 31.9, 38.8, 40.3, 81.0, 98.4, 109.0, 110.7, 111.9, 128.1, 128.7, 130.0, 131.8, 132.4, 132.9, 135.9, 150.5, 162.7; HRMS calcd for C42H41N3O2S3 715.2361, found 715.2350.
1,3-Bis(5-(4-(dimethylamino)phenyl)thiophen-2-yl)-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (4). Using the general procedure, to a solution of aryl halide 12 (100 mg, 0.170 mmol) in DMF under nitrogen atmosphere, PdCl2(PPh3)2 (7.2 mg, 0.01 mmol, 6 mol%), 4-(N,N-dimethylamino)phenylboronic acid 16 (70.1 mg, 0.425 mmol) and K3PO4 (216.2 mg, 1.02 mmol) were added. The title compound 4 was isolated as red solid, 70.1 mg, 61% yield. 1H NMR (400 MHz, CDCl3) δ 0.89 (3H, t, J = 6.6 Hz), 1.24–1.30 (10H, m), 1.66–171 (2H, m), 3.03 (12H, s), 3.68 (2H, t, J = 7.2 Hz), 6.73 (4H, d, J = 9.0 Hz), 7.18 (2H, d, J = 3.9 Hz), 7.55 (4H, d, J = 9.0 Hz), 7.98 (2H, d, J = 4.2 Hz); 13C NMR (100 MHz, CDCl3) δ 14.1, 22.5, 27.0, 28.5, 29.1, 29.6, 31.7, 38.6, 39.7, 111.2, 112.2, 121.9, 125.3, 126.9, 127.4, 128.2, 131.0, 131.4, 134.6, 134.9, 136.6, 152.1, 162.4; HRMS calcd for C38H41N3O2S3 667.2361, found 667.2346.
1,3-Bis(5-(4-(diphenylamino)phenyl)thiophen-2-yl)-5-octyl-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (5). Using the general procedure, to a solution of aryl halide 12 (235 mg, 0.40 mmol) in DMF under nitrogen atmosphere, PdCl2(PPh3)2 (16.9 mg, 0.024 mmol, 6 mol%), 4-(N,N-diphenylamino)phenylboronic acid 17 (289 mg, 1.0 mmol) and K3PO4 (508 mg, 2.40 mmol) were added. The title compound 5 was isolated as red solid, 242.8 mg, 66% yield. 1H NMR (400 MHz, CDCl3) δ 0.90 (3H, t, J = 4.8 Hz), 1.29–1.35 (10H, m), 1.68–1.70 (2H, m), 3.66 (2H, t, J = 7.3 Hz), 7.05–7.16 (16H, m), 7.21 (2H, d, J = 3.9 Hz), 7.28–7.33 (8H, m), 7.50 (4H, d, J = 8.7 Hz), 8.00 (2H, d, J = 4.2 Hz); 13C NMR (100 MHz, CDCl3) δ 14.3, 22.8, 27.2, 28.7, 29.4, 31.7, 31.9, 38.7, 123.1, 123.3, 123.6, 125.0, 126.9, 127.1, 128.0, 129.5, 130.8, 131.3, 136.2, 147.3, 147.7, 148.2, 162.7; HRMS calcd for C58H49N3O2S3 915.2987, found 915.2970.
Acknowledgements
The authors are thankful to Pohang University of Science and technology (POSTECH) for support.
References
- J.-Y. Hu, Y.-J. Pu, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, Adv. Funct. Mater., 2014, 24, 2064 CrossRef CAS PubMed.
- L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137 CrossRef CAS PubMed.
- S. Ren, D. Zeng, H. Zhong, Y. Wang, S. Qian and Q. Fang, J. Phys. Chem. B, 2010, 114, 10374 CrossRef CAS PubMed.
- S. Ellinger, K. R. Graham, P. Shi, R. T. Farley, T. T. Steckler, R. N. Brookins, P. Taranekar, J. Mei, L. A. Padilha, T. R. Ensley, H. Hu, S. Webster, D. J. Hagan, E. W. V. Stryland, K. S. Schanze and J. R. Reynolds, Chem. Mater., 2011, 23, 3805 CrossRef CAS.
- L. Chen, Y. Jiang, H. Nie, R. Hu, H. S. Kwok, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17215 CAS.
- Z. Cai, Y. Guo, S. Yang, Q. Peng, H. Luo, Z. Liu, G. Zhang, Y. Liu and D. Zhang, Chem. Mater., 2013, 25, 471 CrossRef CAS.
- S. S. Dharmapurikar, A. Arulkashmir, C. Das, P. Muddellu and K. Krishnamoorthy, ACS Appl. Mater. Interfaces, 2013, 5, 7086 CAS.
- S. Xu, N. Ai, J. heng, N. Zhao, Z. Lan, L. Wen, X. Wang, J. Peic and X. Wan, RSC Adv., 2015, 5, 8340 RSC.
- Y. Li, G. Zhang, Z. Liu, X. Chen, J. Wang, C. Di and D. Zhang, Macromolecules, 2013, 46, 5504 CrossRef CAS.
- Z. Cai, H. Luo, X. Chen, G. Zhang, Z. Liu and D. Zhang, Chem.–Asian J., 2014, 9, 1068 CrossRef CAS PubMed.
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099 CrossRef CAS PubMed.
- A. Facchetti, Chem. Mater., 2011, 23, 733 CrossRef CAS.
- W. Wu, Y. Liu and D. Zhua, Chem. Soc. Rev., 2010, 39, 1489 RSC.
- S. Steinberger, A. Mishra, E. Reinold, C. M. Muller, C. Uhrich, M. Pfeiffer and P. Bauerle, Org. Lett., 2011, 13, 90 CrossRef CAS PubMed.
- X. Ren, S. Jiang, M. Cha, G. Zhou and Z.-S. Wang, Chem. Mater., 2012, 24, 3493 CrossRef CAS.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638 CrossRef CAS PubMed.
- K. Takimiya, I. Osaka and M. Nakano, Chem. Mater., 2014, 26, 587 CrossRef CAS.
- X. He, B. Cao, T. C. Hauger, M. Kang, S. Gusarov, E. J. Luber and J. M. Buriak, ACS Appl. Mater. Interfaces, 2015, 7, 8188 CAS.
- E. Castro, A. Cabrera-Espinoza, E. Deemer and L. Echegoyen, Eur. J. Org. Chem., 2015, 4629 Search PubMed.
- S. Loser, C. J. Bruns, H. Miyauchi, R. P. Ortiz, A. Facchetti, S. I. Stupp and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 8142 CrossRef CAS PubMed.
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093 CrossRef CAS PubMed.
- X. Liu, Y. Sun, B. B. Y. Hsu, A. Lorbach, L. Qi, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 5697 CrossRef CAS PubMed.
- F. Huang, K.-S. Chen, H.-L. Yip, S. K. Hau, O. Acton, Y. Zhang, J. Luo and A. K.-Y. Jen, J. Am. Chem. Soc., 2009, 131, 13886 CrossRef CAS PubMed.
- P. Shen, H. Bin, L. Xiao and Y. Li, Macromolecules, 2013, 46, 9575 CrossRef CAS.
- K. Li, Z. Li, K. Feng, X. Xu, L. Wang and Q. Peng, J. Am. Chem. Soc., 2013, 135, 13549 CrossRef CAS PubMed.
- B. Kan, Q. Zhang, M. Li, X. Wan, W. Ni, G. Long, Y. Wang, X. Yang, H. Feng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 15529 CrossRef CAS PubMed.
- H.-H. Chang, C.-E. Tsa, Y.-Y. Lai, D.-Y. Chiou, S.-L. Hsu, C.-S. Hsu and Y.-J. Cheng, Macromolecules, 2012, 45, 9282 CrossRef CAS.
- H.-I. Lu, C.-W. Lu, Y.-C. Lee, H.-W. Lin, L.-Y. Lin, F. Lin, J.-H. Chang, C.-I. Wu and K.-T. Wong, Chem. Mater., 2014, 26, 4361 CrossRef CAS.
- A. Mishra, M. K. Fischer and P. Bauerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed.
- C. Teng, X. Yang, C. Yang, H. Tian, S. Li, X. Wang, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2010, 114, 11305 CAS.
- M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235 CrossRef CAS PubMed.
- B. Witulski, T. Schweikert, D. Schollmeyer and N. A. Nemkovich, Chem. Commun., 2010, 46, 2953 RSC.
- R. Fitzner, E. Mena-Osteritz, A. Mishra, G. Schulz, E. Reinold, M. Weil, C. Korner, H. Ziehlke, C. Elschner, K. Leo, M. Riede, M. Pfeiffer, C. Uhrich and P. Bauerle, J. Am. Chem. Soc., 2012, 134, 11064 CrossRef CAS PubMed.
- D. Gajalakshmi, R. V. Solomon, V. Tamilmani, M. Boobalan and P. Venuvanalingam, RSC Adv., 2015, 5, 50353 RSC.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886 CrossRef CAS PubMed.
- J. L. Bredas, R. R. Chance, R. H. Baughman and R. J. Silbey, Chem. Phys., 1982, 76, 3673 CAS.
- J. K. Young and J. S. Moore, Modern Acetylene Chemistry, ed. P. J. Stang and F. Diederich, VCH, Weinheim, Germany, 1995, pp. 415–442 Search PubMed.
- M. B. Nielsen and F. Diederich, Chem. Rev., 2005, 105, 1837 CrossRef CAS PubMed.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS PubMed.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084 RSC.
- A. Hilger, J.-P. Gisselbrecht, R. R. Tykwinski, C. Boudon, M. Schreiber, R. E. Martin, H. P. Luethi, M. Gross and F. Diederich, J. Am. Chem. Soc., 1997, 119, 2069 CrossRef CAS.
- C. R. Swartz, S. R. Parkin, J. E. Bullock, J. E. Anthony, A. C. Mayer and G. G. Malliaras, Org. Lett., 2005, 7, 3163 CrossRef CAS PubMed.
- S. Miao, M. D. Smith and U. H. F. Bunz, Org. Lett., 2006, 8, 757 CrossRef CAS PubMed.
- A. L. Appleton, S. Miao, S. M. Brombosz, N. J. Berger, S. Barlow, S. R. Marder, B. M. Lawrence, K. I. Hardcastle and U. H. F. Bunz, Org. Lett., 2009, 11, 5222 CrossRef CAS PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876 CrossRef CAS PubMed.
- B. D. Rose, D. T. Chase, C. D. Weber, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Org. Lett., 2011, 13, 2106 CrossRef CAS PubMed.
- W. C. W. Leu, A. E. Fritz, K. M. Digianantonio and C. S. Hartley, J. Org. Chem., 2012, 77, 2285 CrossRef CAS PubMed.
- W. C. W. Leu and C. S. Hartley, Org. Lett., 2013, 15, 3762 CrossRef CAS PubMed.
- M. A. Saeed, H. T. M. Le and O. S. Miljanic, Acc. Chem. Res., 2014, 47, 2074 CrossRef CAS PubMed.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, R. A. J. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282 CrossRef CAS PubMed.
- G. Jayamurugan, O. Dumele, J.-P. Gisselbrecht, C. Boudon, W. B. Schweizer, B. Bernet and F. Diederich, J. Am. Chem. Soc., 2013, 135, 3599 CrossRef CAS PubMed.
- B. Liu, W. Zhu, Y. Wang, W. Wu, X. Li, B. Chen, Y.-T. Long and Y. Xie, J. Mater. Chem., 2012, 22, 7434 RSC.
- I. Kaur, W. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274 CrossRef CAS PubMed.
- O. L. Griffith, J. E. Anthony, A. G. Jones, Y. Shu and D. L. Lichtenberger, J. Am. Chem. Soc., 2012, 134, 14185 CrossRef CAS PubMed.
- Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039 RSC.
- B. C. Tlach, A. L. Tomlinson, A. Bhuwalka and M. Jeffries-EL, J. Org. Chem., 2011, 76, 8670 CrossRef CAS PubMed.
- Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799 CrossRef CAS PubMed.
- L. Zophel, V. Enkelmann and K. Mullen, Org. Lett., 2013, 15, 804 CrossRef PubMed.
- V. Bhalla, G. Singh, M. Kumar, C. Singh, M. Rawat and R. S. Anand, RSC Adv., 2013, 3, 14722 RSC.
- A. L. Korich, I. A. McBee, J. C. Bennion, J. I. Gifford and T. S. Hughes, J. Org. Chem., 2014, 79, 1594 CrossRef CAS PubMed.
- A. Najari, S. Beaupre, P. Berrouard, Y. Zou, J.-R. Pouliot, C. Lepage-Perusse and M. Leclerc, Adv. Funct. Mater., 2011, 21, 718 CrossRef CAS PubMed.
- P. Berrouard, A. Najari, A. Pron, D. Gendron, P.-O. Morin, J.-R. Pouliot, J. Veilleux and M. Leclerc, Angew. Chem., Int. Ed., 2012, 51, 2068 CrossRef CAS PubMed.
- A. Najari, P. Berrouard, C. Ottone, M. Boivin, Y. Zou, D. Gendron, W.-O. Caron, P. Legros, C. N. Allen, S. Sadki and M. Leclerc, Macromolecules, 2012, 45, 1833 CrossRef CAS.
- H. Zhong, Z. Li, F. Deledalle, E. C. Fregoso, M. Shahid, Z. Fei, C. B. Nielsen, N. Yaacobi-Gross, S. Rossbauer, T. D. Anthopoulos, J. R. Durrant and M. Heeney, J. Am. Chem. Soc., 2013, 135, 2040 CrossRef CAS PubMed.
- J. Yuan, Z. Zhai, H. Dong, J. Li, Z. Jiang, Y. Li and W. Ma, Adv. Funct. Mater., 2013, 23, 885 CrossRef CAS PubMed.
- Q. Wu, M. Wang, X. Qiao, Y. Xiong, Y. Huang, X. Gao and H. Li, Macromolecules, 2013, 46, 3887 CrossRef CAS.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupre, B. R. Aich, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330 CrossRef CAS PubMed.
- T.-Y. Chu, J. Lu, S. Beaupre, Y. Zhang, J.-R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250 CrossRef CAS PubMed.
- C. Cabanetos, A. E. Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Frechet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656 CrossRef CAS PubMed.
- G.-Y. Chen, Y.-H. Cheng, Y.-J. Chou, M.-S. Su, C.-M. Chen and K.-H. Wei, Chem. Commun., 2011, 47, 5064 RSC.
- Q. Feng, X. Lu, G. Zhou and Z.-S. Wang, Phys. Chem. Chem. Phys., 2012, 14, 7993 RSC.
- Q. Zhang, H. Kuwabara, W. J. Potscavage Jr, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070 CrossRef CAS PubMed.
- Z. R. Grabowksi, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899 CrossRef PubMed.
- G. M. Badger and I. S. Walker, J. Chem. Soc., 1956, 122 RSC.
- M. J. Frisch, et al., Gaussian 03, revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Normalized UV-visible and fluorescence spectra, fluorescence images of the dyes, cyclic voltammograms, Cartesian coordinates, NMR spectra. See DOI: 10.1039/c5ra13416a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.