DOI:
10.1039/C5RA13344K
(Communication)
RSC Adv., 2015,
5, 76079-76082
High-yielding and facile synthesis of organosilicon compounds containing a m-carboranylmethyl group†
Received
8th July 2015
, Accepted 4th September 2015
First published on 4th September 2015
Abstract
An efficient method for the synthesis of organosilicon compounds containing m-carboranylmethyl was developed, which afforded the products in good to excellent yields (up to 88%) compared to the literature methods affording a 38% yield. Moreover, the generated intermediate 5 containing a Si–Br bond could be functionalized conveniently.
Icosahedral carboranes are an interesting class of exceptionally stable boron-rich clusters that have attracted significant attention due to their broad range of potential applications. For instance, they are used as key building blocks in the synthesis of complex polymeric molecules,1 in materials and organometallic chemistry,2 and as boron neutron capture therapy agents in the pharmaceutical field.3 Since the discovery of carboranes in 1960s, they have been incorporated into polysiloxanes, excellent heat resistant materials, which exhibited significant improvement in the thermal and oxidative stability thereof.4 Compared to the m-carborane polymer a, polymer b exhibited higher stability towards both nucleophilic and electrophilic reagents; meanwhile, it showed higher thermal oxidation behavior compared to polymer c (Fig. 1).
 |
| | Fig. 1 m-Carborane-containing polysiloxanes. | |
This could be attributed to the fact that m-carboranylsilyl polymer b in which the carborane units are separated from the silicon atoms by one methylene bridge is more stable and promising from the practical perspective.5 An increase in the number of methylene groups reduces the resistance to degradation. Nonetheless, there are only a few reports about the synthesis of the m-carboranylmethylsilyl compounds, and the reported yields of the desired compounds are too low (38%), thus indicating difficulty of derivatization (Scheme 1).6 Therefore, exploration of a high-yielding, mild, efficient, and facile method for the synthesis of the m-carboranylmethylsilyl compounds is still a challenging task.
 |
| | Scheme 1 Previous routes for the synthesis of m-carboranylmethylsilyl compounds. | |
According to the literature (Scheme 1: 1981), the yield of the desired product I (19.1%) was lower than that of II (57.3%), thus indicating that in the chloromethyl(organo)alkoxysilane compounds, the reactivity of Si–O bond is higher than that of C–Cl bond towards nucleophilic substitution reaction, leading to extremely low yield of the desired products, i.e., m-carboranylmethylsilyl compounds. Inspired by the protective groups of the organic chemistry, we developed a novel and efficient method for the synthesis of derivatives of silylmethyl m-carborane utilizing the protection and deprotection strategy (Fig. 2).
 |
| | Fig. 2 Retrosynthetic analysis. | |
We commenced our study using phenyl as protecting group, and screened the solvent and temperature initially (Table 1) to incorporate silylmethyl group into m-carborane. First, diethyl ether (Et2O) was selected as the solvent of choice; however, the reaction did not start even at 35 °C. Further, when the reaction was performed at 67 °C using tetrahydrofuran (THF) as the solvent instead of Et2O, the yield of compound 1 was 11%. Thus, we could infer that temperature was one of the important factors. Subsequently, dimethoxyethane (DME), dibutyl ether (DE), and anisole (PhOMe) were screened as reaction solvent, respectively. Simultaneously, the effect of increasing the reaction temperature was also investigated; however, the yield of compound 1 was still low. It was supposed that linear ether solvents were not favorable for this reaction because of their weak chelation effect, which reduced the nucleophilicity of lithiated carborane. Hence, we selected the high boil point (88 °C) cyclic ether tetrahydropyran (THP) as the reaction solvent and optimized the lithiation temperature. Consequently, the yield of 1 could reach up to 57%, which was higher than that the lithiation solvent was dimethoxyethane (27%). It was concluded that reaction temperature and solvent played the most important roles. THP was the optimized solvent, and room temperature was favorable for the lithiation reaction. The entries 1–5 afforded oily liquids; however, 6–8 exhibited the mixture of oily liquid and white solid. Unfortunately, it was extremely difficult to separate the products 1 and 2 by either column chromatography or recrystallization attributed to their fairly similar polarity and solubility (Scheme 2).
Table 1 Screening of solvent and temperaturea
| Entry |
Solventb |
Lithiation temperature (°C) |
Reaction temperature (°C) |
1 yieldc (%) |
| The reaction was conducted on 5 mmol carborane in 20 mL solvent. THP = tetrahydropyran, DME = dimethoxyethane, DE = dibutyl ether. Determined by GC. |
| 1 |
Et2O |
0 |
35 |
— |
| 2 |
THF |
0 |
67 |
11 |
| 3 |
DME |
0 |
90 |
27 |
| 4 |
Dioxane |
0 |
100 |
24 |
| 5 |
PhOMe |
0 |
145 |
30 |
| 6 |
DE |
0 |
150 |
32 |
| 7 |
THP |
0 |
95 |
41 |
| 8 |
THP |
0 to rt |
95 |
46 |
| 9 |
THP |
rt |
95 |
57 |
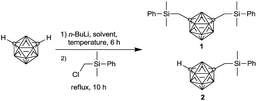 |
| | Scheme 2 Synthesis route for the preparation of m-carboranylmethylsilyl compound with phenyl protecting group. | |
Therefore, it was assumed that monosubstituted and bis-substituted m-carboranes could be separated easily by increasing the polarity of the aryl group, thus; the protecting group was changed to p-methoxyphenyl (PMP). Consequently, 1,7-bis(4-methoxyphenyl(dimethyl)silylmethyl)-m-carborane 3 (molecular structure identified by X-ray crystallography is shown in Fig. 3) was first synthesized easily and conveniently, and obtained as a mixture containing monosubstitued m-carborane 4 (Scheme 3). As might be expected, 3 was isolated successfully by recrystallization. Simultaneously, lithiation time was optimized by performing the reaction for 0.5, 1, 3, and 6 h, respectively (Table 2). The results indicated that the yield was the maximum when the lithiation reaction was performed for 1 h. Finally, 3 was obtained in 50% yield after recrystallization, and the GC yield was 83%, as well as 7% of 4 was obtained. When the recrystallization solution containing 3 and 4 was further treated with n-BuLi and (chloromethyl) (4-methoxyphenyl)dimethylsilane, we found that 4 was transformed into 3. Therefore, the overall yield reached up to 88% which outclassed the results of the previous study, i.e., 38%.
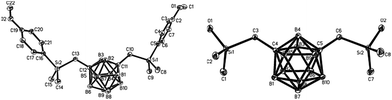 |
| | Fig. 3 The molecular structure of 3 (left) and 6 (right).7 | |
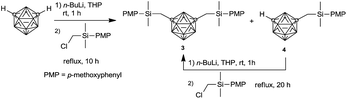 |
| | Scheme 3 Synthesis route of m-carboranylmethylsilyl compound with PMP protecting group. | |
Table 2 Screening of the lithiation time
| Determined by GC. |
| Lithiation time (h) |
0.5 |
1 |
3 |
6 |
| 3 yield (%)* |
77 |
83 |
77 |
59 |
Subsequently, we focused on the deprotection and functionalization of 3. Initially, HCl, H2SO4, TfOH, HBF4·H2O, and KOH were used as deprotecting agents, respectively. However, deprotection using HCl, H2SO4, TfOH, and HBF4·H2O resulted in the formation of complex mixture of the products, which was difficult to purify. Furthermore, the reaction did not start when KOH was used as deprotecting agent, thus success was not achieved. It is well-known that Br2 is a strong electrophile which was utilized to cleave the aryl-Si bond.8 Therefore, the Si–PMP bond of 3 was cleaved successfully to generate 1,7-bis(bromo(dimethyl)silylmethyl)-m-carborane 5 (Scheme 4). Notably, the byproducts p-bromoanisoles could be recycled and utilized to synthesize (chloromethyl)(4-methoxyphenyl)dimethylsilane. In virtue of high reactivity of Si–Br bond, 5 would be a very useful intermediate to achieve the functionalization of silylmethyl-m-carborane, for instance, 5 could conveniently undergo extensive range of reactions with various nucleophiles such as H2O, MeOH, Et2NH, and Grignard reagent (Fig. 4); or be reduced to Si–H.9 Therefore, in this study, we focused on the hydrolysis and reduction of 5.
 |
| | Scheme 4 Deprotection of 3. | |
 |
| | Fig. 4 Derivatization of 5. | |
5 was extremely easy to hydrolyze and the byproduct HBr was the promoter of the self-condensation of hydrolysate 6; therefore, we screened the absorbent for HBr, hydrolytic temperature, and operational approach. Finally, 1,7-bis(hydroxy(dimethyl)silylmethyl)-m-carborane 6 (the structure is shown in Fig. 3) was obtained nearly quantitatively after recrystallization with Et3N used as absorbent at −15 °C (Scheme 5). The operation details are provided in the ESI.†
 |
| | Scheme 5 Derivatization of 5. | |
5 was successfully reduced quantitatively to 1,7-bis(dimethylsilylmethyl)-m-carborane 7 using LiAlH4 as reducing agent (Scheme 5). Similarly, 1,7-bis(vinyldimethylsilylmethyl)-m-carborane 8 and 1,7-bis(ethynyldimethylsilylmethyl)-m-carborane 9 were synthesized through Grignard reaction.
In summary, a novel and facile synthetic method utilizing p-methoxyphenyl as protecting group and Br2 as deprotecting reagent was developed to prepare functional organosilicon compounds containing m-carboranylmethyl group. The yield reached up to 88%. Furthermore, four significantly important derivatives, namely, 1,7-bis(hydroxy(dimethyl)silylmethyl)-m-carborane 6, 1,7-bis-((dimethyl)silylmethyl)-m-carborane 7, 1,7-bis(vinyldimethylsilylmethyl)-m-carborane 8, and 1,7-bis(ethynyldimethylsilylmethyl)-m-carborane 9 were synthesized in high yield. The intermediate 1,7-bis(bromo(dimethyl)silylmethyl)-m-carborane 5 conveniently afforded the difunctional silylmethyl-m-carborane compounds. The method was environmentally friendly because the byproduct 4-bromoanisole could be recycled. Moreover, the prominent advantage of the method was the simple and convenient method of purification; i.e., recrystallization, thus paving the way for large-scale preparation. Furthermore, this method could contribute significantly to provide a scientific breakthrough and enrich the functionalization of m-carborane as well as lay a solid foundation for the application of m-carborane.
Notes and references
- For selected examples, see:
(a) X. Huang, Q. Zhang, Z. Meng, J. Gu, X. Jia and K. Xi, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 973–980 CrossRef CAS PubMed;
(b) Y. Jiang, X. Li, F. Huang, Y. Zhou and L. Du, J. Macromol. Sci., Part A: Pure Appl. Chem., 2015, 52, 476–484 CrossRef CAS PubMed;
(c) K. Kokado, Y. Tokoro and Y. Chujo, Macromolecules, 2009, 42, 2925–2930 CrossRef CAS;
(d) X. Zhang, L. Kong, L. Dai, X. Zhang, Q. Wang, Y. Tan and Z. Zhang, Polymer, 2011, 52, 4777–4784 CrossRef CAS PubMed;
(e) J. Marshall, J. Hooton, Y. Han, A. Creamer, R. S. Ashraf, Y. Porte, T. D. Anthopoulos, P. N. Stavrinou, M. A. McLachlan, H. Bronstein, P. Beavise and M. Heeney, Polym. Chem., 2014, 5, 6190–6199 RSC;
(f) T. Xing, Y. Huang, K. Zhang and J. Wu, RSC Adv., 2014, 4, 53628–53633 RSC.
- For selected examples, see:
(a) D. Zhao, J. Zhang and Z. Xie, Angew. Chem., Int. Ed., 2014, 53, 8488–8491 CrossRef CAS PubMed;
(b) Y. Quan and Z. Xie, J. Am. Chem. Soc., 2014, 136, 15513–15516 CrossRef CAS PubMed;
(c) H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., Int. Ed., 2015, 54, 5084–5087 CrossRef CAS PubMed;
(d) Y. Quan, Z. Qiu and Z. Xie, J. Am. Chem. Soc., 2014, 136, 7599–7602 CrossRef CAS PubMed;
(e) Z. Qiu, Tetrahedron Lett., 2015, 56, 963–971 CrossRef CAS PubMed;
(f) L. Zhu, X. Tang, Q. Yu, W. Lv, H. Yan, Q. Zhao and W. Huang, Chem.–Eur. J., 2015, 21, 4721–4730 CrossRef CAS PubMed;
(g) S. Ren, Z. Qiu and Z. Xie, Angew. Chem., Int. Ed., 2012, 51, 1010–1013 CrossRef CAS PubMed;
(h) M. Koshino, T. Tanaka, N. Solin, K. Suenaga, H. Isobe and E. Nakamura, Science, 2007, 316, 853–854 CrossRef CAS PubMed;
(i) B. P. Dash, R. Satapathy, J. A. Maguireb and N. S. Hosmane, New J. Chem., 2011, 35, 1955–1972 RSC;
(j) J. U. Kahlert, A. Rawal, J. M. Hook, L. M. Rendina and M. Choucair, Chem. Commun., 2014, 50, 11332–11334 RSC.
- For selected reviews, see:
(a) A. H. Soloway, W. Tjarks, B. A. Barnum, F.-G. Rong, R. F. Barth, I. M. Codogni and J. G. Wilson, Chem. Rev., 1998, 98, 1515–1562 CrossRef CAS PubMed;
(b) J. Plesek, Chem. Rev., 1992, 92, 269–278 CrossRef CAS;
(c) M. F. Hawthorne, Angew. Chem., Int. Ed., 1993, 32, 950–984 CrossRef PubMed;
(d) R. N. Grimes, Dalton Trans., 2015, 44, 5939–5956 RSC.
-
(a) E. N. Peters, J. Macromol. Sci., Polym. Rev., 1979, 17, 173–208 CrossRef PubMed;
(b) E. N. Peters, Ind. Eng. Chem. Prod. Res. Dev., 1984, 23, 28–32 CrossRef CAS.
- N. J. Bekasova and N. G. Komarova, Russ. Chem. Rev., 1992, 61, 352–362 CrossRef PubMed.
-
(a) V. N. Kalinin, B. A. Izmailov, A. A. Kazantsev, V. D. Myakushev, A. A. Zhdanov and L. I. Zakharkin, J. Organomet. Chem., 1981, 216, 295–320 CrossRef CAS;
(b) B. A. Izmailov, V. N. Kalinin, V. D. Myakushev, A. A. Zhdanov and L. A. Zakharkin, Dokl. Akad. Nauk SSSR, 1985, 280, 114–118 CAS;
(c) B. A. Izmailov, V. I. Nedel'kin and I. S. Gerr, Russ. Chem. Bull., 1998, 47, 687–690 CrossRef CAS.
- ESI†.
- C. Eaborn, J. Organomet. Chem., 1975, 100, 43–57 CrossRef CAS.
-
(a) C. Eaborn, Organosilicon Compounds, Academic Press, New York, 1960, ch. 5, p. 167 Search PubMed;
(b) C. Eaborn and M. N. Romanelli, J. Chem. Soc., Perkin Trans. 2, 1987, 657–662 RSC;
(c) D. Azarifar, M. P. Coles, S. M. El-Hamruni, C. Eaborn, P. B. Hitchcock and J. D. Smith, J. Organomet. Chem., 2004, 689, 1718–1722 CrossRef CAS PubMed;
(d) J. A. Hartsel, D. M. Wong, J. M. Mutunga, M. Ma, T. D. Anderson, A. Wysinski, R. Islam, E. A. Wong, S. L. Paulson, J. Li, P. C. H. Lam, M. M. Totrov, J. R. Bloomquist and P. R. Carlier, Bioorg. Med. Chem. Lett., 2012, 22, 4593–4598 CrossRef CAS PubMed;
(e) C. Eaborn and P. D. Lickiss, J. Organomet. Chem., 1985, 294, 305–313 CrossRef CAS;
(f) J. Barrau, N. B. Hamida, A. Agrebi and J. Satge, Organometallics, 1989, 8, 1585–1593 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1404643 and 1404644. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra13344k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 








