
Synthesis of flower-shaped ZrO2–C composites for adsorptive removal of trichlorophenol from aqueous solution
Yixin Tanabc,
Lehui Zhua,
Hongyun Niu*b,
Yaqi Caib,
Fengchang Wuc and
Xiaoli Zhao*c
aDepartment of Resources Environmental and Chemical Engineering of Nanchang University, Jiangxi, Nanchang Province 330031, China
bState Key Laboratory of Environmental Chemistry and Ecotoxicology of Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, P. O. Box 2871, Beijing 100085, China. E-mail: hyniu@rcees.ac.cn; Fax: +86-010-62849182; Tel: +86-010-62849182
cState Key Laboratory of Environmental Criteria and Risk Assessment, Chinese Research Academy of Environmental Sciences, Beijing, 100012, China
First published on 2nd September 2015
Abstract
In the current study, examples of a novel kind of nanoflake zirconia–carbon (ZrO2–C) composite were synthesized through a simple method by using gallic acid and ZrCl4 as precursors. The as-synthesized ZrO2–C composites were observed to have high specific surface areas and a chrysanthemum-like structure. High-resolution transmission electron microscopy, X-ray photoelectron spectroscopy, infrared spectroscopy, X-ray diffraction, and Raman analyses revealed that the ZrO2–C composites were composed of graphitized carbon and numerous ZrO2 nanoparticles (3–4 nm in diameter). The ZrO2–C composites were successfully used as adsorption materials to remove 2,4,6-trichlorophenol (TCP) from simulated water samples. The results showed that ZrO2–C exhibited a much higher adsorption capacity for TCP than did some reported carbon materials. The hydrophobic interaction and/or π–π stacking interaction between TCP and the carbon phase, hydrogen bonding with functional groups of ZrO2–C, and metal–anion binding with ZrO2 nanoparticles contributed to the high adsorption ability. Generally, TCP uptake was favorable in an acidic environment and increased as the initial TCP concentration and temperature were increased. The adsorption process obeyed pseudo-second-order kinetics, and the adsorption isotherms could be well described by the Freundlich equation.
1. Introduction
Chlorophenols are widely used in petroleum refining, plastics, rubbers, pharmaceuticals, disinfectants, wood preservation, steel industries, and the pulp and paper industry. Due to their carcinogenic and mutagenic nature and high resistance to biodegradation, chlorophenol pollutants are now the subject of great concern.1,2 Much effort has been expended to physicochemically (such as by adsorption,1–14 ozonation,15–17 and electrochemical oxidation18) and biologically treat chlorophenol-rich wastewater.19,20Over the last century, much research has been conducted in the area of removal of chlorophenols by carbon materials (including activated carbon, carbon nanotubes, graphene, and mesoporous carbon) which have been shown to display high efficiency at removing organic pollutants.1–10 Soil, metal oxides and zeolite have been explored as adsorbents to remove chlorophenols from aqueous solutions as well.11–14,21–23 The sorption of chlorophenols on these inorganic adsorbents were indicated to proceed via different reaction mechanisms, including inner- or outer-sphere coordination, H-bonding, ligand exchange between the OH groups of phenolic compounds and the active sites (hydroxyl groups) of adsorbents, and complexation of phenolate anions with metal ions on the surface of adsorbents. Yousef et al. showed an adsorption capacity of chlorophenols onto natural zeolites as high as 0.32 mmol g−1, which was comparable with those obtained by some carbon materials.11 Therefore, metal oxides, soil or zeolites can be regarded as potential adsorbents to remove chlorophenols from environmental waters.
Herein, we propose a kind of combination of carbon materials and metal oxides that can provide multiple adsorptive sites for chlorophenols and that can interact with the chlorophenols by different mechanisms. Gallic acid (GA), a phenolic compound belonging to the hydroxybenzoic acids, is present in high quantities in legumes (onions, black radish, hops), fruits (grapes, some red fruits), nuts (gallnuts) and beverages (wine, beer). GA can form a complex with Fe(II), Fe(III), Cu(II), Al(III), Zr(IV), Mo(VI), and other metal ions.24–28 GA has been used in the green synthesis of AgNPs and AuNPs as well.29 However, to the best of our knowledge, GA has not been utilized as an organic ligand in the synthesis of hierarchically structured nanoparticles. In the current study, we used GA as both an organic ligand and carbon precursor to prepare carbon and metal oxide composites. Zirconium oxides were used as metal precursors due to the excellent chemical stability of Zr-based inorganic or organic materials.30 The method used to synthesize ZrO2–C composites is very simple and produced high yields. The as-prepared ZrO2–C composites were observed to display a chrysanthemum-like structure and were used as adsorbents to remove 2,4,6-trichlorophenol from water.
2. Experimental section
2.1 Chemicals and materials
Zirconium tetrachloride, hydrochloric acid (HCl, 36–38%), ethanol, and N,N-dimethylformamide (DMF, 99.5%) were purchased from Sinopharm Chemistry Reagent Co., Ltd (Beijing, China). 2,4,6-Trichlorophenol (TCP) and gallic acid were obtained from J&K Chemical Ltd (Beijing, China). TCP (100 mg L−1) was prepared and diluted to a specified concentration when necessary. Ultrapure water used in all of the experiments was prepared by using the Milli-Q SP reagent water system (Millipore, Bedford, MA, USA).2.2 Synthesis and structure of ZrO2–C
First, ZrCl4 (0.933 g) was dissolved in 5 mL of ethanol, and then mixed with 65 mL of DMF, followed by the addition of 0.753 g of gallic acid to the mixture under stirring. The reaction solution was transferred into a Teflon-lined stainless-steel autoclave, which was sealed to heat the solution at 403 K for 24 h. The white products were washed with DMF three times and soaked in ethanol for three days (with ethanol changed each day), and then dried at 323 K in a vacuum environment for 12 h. Finally, the product (1.348 g) was removed to a quartz tube to be carbonized under the protection of nitrogen at 1073 K for 4 h.2.3 Characterization of the material
The sizes and morphologies of the synthesized materials were surveyed using a Hitachi S-5500 field-emission scanning electron microscope (FE-SEM, Tokyo, Japan) equipped with an energy dispersive X-ray spectrometer (SEMEDX, Tokyo, Japan), a JEOL JEM-2010 high-resolution transmission electron microscope (HRTEM, Kyoto, Japan), and a Hitachi H-7500 transmission electron microscope (TEM, Tokyo, Japan). The crystalline phase of each product was analyzed using X-ray powder diffraction (XRD, Almelo, Netherlands) by using Cu Kalpha radiation ranging from 5° to 70° with a resolution of 0.02°. Brunauer–Emmett–Teller (BET) methods were adopted to measure the surface area, pore size and volume of the product (ASAP2000 V3.01A; Micromeritics, Norcross, GA). Fourier transform infrared spectroscopy (FTIR) spectra were recorded in the range of 4000–400 cm−1 by a NEXUS 670 infrared Fourier transform spectrometer (Nicolet Thermo, Waltham, MA) after pelletizing with KBr. Thermogravimetric analysis (TGA) for freeze-dried samples were carried out on a Mettler Toledo Star TGA/SDTA 851 apparatus, and the temperature was increased at a rate of 10 K min−1 from room temperature to 1173 K. The sample chamber was purged with dry nitrogen. To detect the composition and chemical state of elements on the surface of the materials, the products were analyzed using X-ray photoelectron spectroscopy (XPS) with an ESCA-Lab-200i-XL spectrometer (Thermo Scientific, Waltham, MA) using monochromatic Al Kalpha radiation (1486.6 eV).2.4 Absorptive removal of pollutants
Absorption experiments were conducted in simulated polluted water (10 mL) as batch experiments. The concentration of adsorbent was 0.02 g L−1. The solution pH was adjusted with HCl and NaOH to desired values, and the ionic strength was adjusted with 1 M NaCl. The effect of initial pH on the sorption of TCP was studied in the pH range of 3.0–10.0. Dynamic experiments were operated at a temperature of 303 K and using 20 mg L−1 TCP. The influence of ionic strength was tested by adding NaCl (10, 20, and 30 mM) into the solution. To investigate the thermodynamic properties, adsorption isotherms were recorded in 10 mL of TCP solution with concentrations of TCP varying from 0.5 to 20 mg L−1 at 303 K, 313 K, and 323 K, respectively. All experiments were conducted in triplicate, and average results were reported.We calculated the maximum and equilibrium adsorption levels by measuring the concentration of the original TCP and of the equilibrium TCP throughout the whole process. The adsorption formula used was
 | (1) |
The dosage of TCP was determined by using an HPLC system equipped with a DIONEX HPLC pump (P680), a thermostatted column compartment (TCC-100) and a photodiode array detector (PDA-100). Separations were conducted on a Dikma C18 column (250 × 4.6 mm; 5 μm). The mobile phase for TCP was acetonitrile: 2% HAc (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v) at a flow rate of 1 mL min−1. The wavelength was set at 290 nm.
:20 v/v) at a flow rate of 1 mL min−1. The wavelength was set at 290 nm.
3. Results and discussion
3.1 Characterization and properties of adsorbents
Fig. 1 shows the TEM and SEM images of the as-prepared materials. The Zr–GA particles displayed a chrysanthemum-like shape with dimensions of about 4 μm (Fig. 1A and B). This material was observed to be composed of nanoflakes (Fig. 1C) ∼350 nm in width in the high-resolution TEM image of Zr–GA. After carbonization, the chrysanthemum-like shape of the ZrO2–C composites remained, but the particle size decreased to about 2 μm (Fig. 1D and E). The Zr–GA nanoflakes were observed to be reduced to about 100–300 nm in width and to contain numerous tiny (3–4 nm in diameter) nanoparticles that were presumably ZrO2 (Fig. 1F and G). | ||
| Fig. 1 SEM images of Zr–GA (A) and ZrO2–C (D), TEM images of Zr–GA (B and C) and ZrO2–C (E and F), and an HRTEM image of ZrO2–C (G). | ||
The crystal phases of the samples were investigated by XRD analysis. Fig. 2 shows that Zr–GA gave a totally different XRD pattern than did ZrO2 and GA powder; its diffraction peaks occurring at 7.2°, 9.0°, 11.3°, 16.8°, 17.3°, 21.3°, 23.5°, 26.3°, 28.5°, 31.1°, and 36.6° basically conformed to Zr–MOFs, which are synthesized by zirconium metal salts and organic compounds with carboxyl groups in other studies.31–34 Since the organic ligand (gallic acid) used in our study differed from the traditional organic ligands used to synthesize MOFs, small deviations of some of the peaks may occur. The characteristic peaks suggest the chelation of Zr atoms with GA in Zr–GA. In the XRD pattern of ZrO2–C, the distinguishing characteristic peaks appeared at 2θ = 30.1°, 35.4°, 50.2°, and 60.2°, which can be attributed to the (101), (002), (112), and (211) diffraction planes of the tetragonal zirconia.35,36
 | ||
| Fig. 2 XRD spectra of GA, Zr–GA and ZrO2–C from 2θ = 5 to 70°. | ||
Fig. 3A shows the IR spectra of GA, Zr–GA and ZrO2–C. In the spectrum of GA, the broad peaks at 3368 cm−1 and 3287 cm−1 correspond to the phenolic groups. The peaks ranging from 3200–2500 cm−1 are associated with the bending or stretching vibrations of carboxylic acid O–H and of C–H in aromatic groups. The sharp peak at 1701 cm−1 is due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration from carboxylic groups. The peaks in the region of 1500–1000 cm−1 were assigned to the stretching vibration of C–O, and to the binding, upon adsorption, of the O–H bonds of carboxylic and phenolic groups and of the C–H bonds of the aromatic rings (Fig. 3B). After interaction with Zr(IV), the O–H stretching band in the 3400–2500 cm−1 region, the Zr–O–H bending peaks at 1332 cm−1, and the carboxylic group O–H bending band at 1029 cm−1 disappeared, indicating that all the phenolic groups and carboxylic groups in GA chelated with Zr. Correspondingly, the CO stretching band in the Zr–GA spectrum shifted to a low frequency, and two characteristic carboxylate group (COO−) bands between 1300 and 1600 cm−1 appeared. Asymmetrical stretching of the carboxylate groups occurred at 1500 cm−1 with stretching at 1426 cm−1.37 These results indicate that all the carboxylic and phenolic groups in the molecules interacted with Zr. Actually, the carboxylic groups are vital for the formation of the chrysanthemum-like structure of these materials. When tannic acid was adopted as a linker, the hierarchical morphology of the as-prepared products could not be observed. In the ZrO2–C spectrum, the characteristic peaks for carboxylic groups, phenolic groups and benzene rings generally disappeared, suggesting the successful carbonization of GA; the broad peaks in the 3600–3300 cm−1 and 1670–1550 cm−1 regions resulted from the surface-adsorbed water and hydroxyl groups of the carbon materials.
O stretching vibration from carboxylic groups. The peaks in the region of 1500–1000 cm−1 were assigned to the stretching vibration of C–O, and to the binding, upon adsorption, of the O–H bonds of carboxylic and phenolic groups and of the C–H bonds of the aromatic rings (Fig. 3B). After interaction with Zr(IV), the O–H stretching band in the 3400–2500 cm−1 region, the Zr–O–H bending peaks at 1332 cm−1, and the carboxylic group O–H bending band at 1029 cm−1 disappeared, indicating that all the phenolic groups and carboxylic groups in GA chelated with Zr. Correspondingly, the CO stretching band in the Zr–GA spectrum shifted to a low frequency, and two characteristic carboxylate group (COO−) bands between 1300 and 1600 cm−1 appeared. Asymmetrical stretching of the carboxylate groups occurred at 1500 cm−1 with stretching at 1426 cm−1.37 These results indicate that all the carboxylic and phenolic groups in the molecules interacted with Zr. Actually, the carboxylic groups are vital for the formation of the chrysanthemum-like structure of these materials. When tannic acid was adopted as a linker, the hierarchical morphology of the as-prepared products could not be observed. In the ZrO2–C spectrum, the characteristic peaks for carboxylic groups, phenolic groups and benzene rings generally disappeared, suggesting the successful carbonization of GA; the broad peaks in the 3600–3300 cm−1 and 1670–1550 cm−1 regions resulted from the surface-adsorbed water and hydroxyl groups of the carbon materials.
 | ||
| Fig. 3 FTIR spectra of GA, Zr–GA and ZrO2–C (A) and FTIR spectra of GA and Zr–GA in the 400–1800 cm−1 region (B). | ||
XPS was used to investigate the elemental composition on the surface of Zr–GA before and after carbonization. Curve fitting of the O1s and Zr3d lines for both materials employed Gaussian (20%)–Lorentzian (80%) peak shapes, respectively (defined in Casa XPS as GL (80)). The O1s core levels for Zr–GA showed three main components related to C–OH, COO− and H2O species (Fig. 4A). Zr–GA gave rise to Zr3d spectra characterized by Zr3d3/2 and Zr3d5/2 doublet terms due to spin–orbit coupling, and the positions of Zr3d5/2 and Zr3d3/2 peaks located at 182.5 and 185.0 eV (Fig. 4B), respectively, which is consistent with the zirconium(IV) cations coordinated with carboxylic oxygens of organic ligands in the zirconium metal–organic framework and the amine-functionalized zirconium metal–organic framework.38 After carbonization, the O1s core level shifted to lower binding energies with the disappearance of H2O species and significant decrease in the amounts of C–OH and COO− species; meanwhile, the Zr–O bond appeared. The intensity of the ZrO2–C Zr3d spectrum increased obviously, and the positions of Zr3d peaks were observed to shift to lower binding energies (182.4 and 184.8 eV, respectively). The Zr3d spectra of Zr–GA and ZrO2–C were generally observed to be in good agreement with the reported values for Zr(IV) cations. Wang39 and co-workers found the binding energy of the Zr3d in hybrid ZrO2/polymer nanoparticles to be higher than that in ZrO2, suggesting the formation of chemical bonds between the Zr(IV) and the organic components. Similar to the case of the zirconium metal–organic framework, we deduced that the Zr atoms coordinated the oxygen atoms from gallic acids in the chrysanthemum-like Zr–GA structure, while zirconium transformed to ZrO2 during carbonization. In the ZrO2–C Raman spectrum (Fig. 4C), two peaks were observed to be centered at 1580 cm−1 (G-line) and 1350 cm−1 (D-line), corresponding to in-plane vibrations of crystalline graphite and disordered amorphous carbon, respectively, and suggesting that the GA linkers were successfully graphitized. The above-mentioned results imply that ZrO2–C is composed of ZrO2 and graphitized carbon.
 | ||
| Fig. 4 The fitted XPS O1s spectra (A) and Zr3d core level lines (B) of Zr–GA and ZrO2–C. Raman spectra (C), TGA curves (D) and N2 adsorption/desorption isotherm (E) of ZrO2–C. Inset in (E) describes the calculated pore size distribution of ZrO2–C. | ||
According to TGA (Fig. 4D), ZrO2–C underwent a slight weight decrease, due to the initial loss of water, upon being heated up to 473 K. Upon being heated to above 623 K, the carbon phase of the ZrO2–C decomposed promptly, suggesting the oxidation of carbon. The percent remaining after 713 K can be regarded as pure ZrO2–C. The percentage of carbon in the mass of ZrO2–C was estimated from the TGA results to be 40%. The N2 adsorption/desorption isotherm and calculated pore size distribution of ZrO2–C (Fig. 4E) suggests that the adsorption process conformed to the IV-type with H3-type hysteresis loops, indicative of mesoporous pores. The total pore volume of ZrO2–C was determined to be 0.25 cm3 g−1. The average pore size calculated from desorption branch of the N2 isotherm by the Barrett–Joyner–Halenda (BJH) method was 3.8 nm, and the Brunauer–Emmett–Teller (BET) surface area of the obtained ZrO2–C composite was 79.4 m2 g−1.
3.2 Adsorption kinetics
The kinetics of TCP adsorption onto ZrO2–C was analyzed using both pseudo-first-order and pseudo-second order kinetic models. The pseudo-first-order kinetic model is defined as| In(qe − qt) = Inqe − k1t | (2) |
The pseudo-second-order constants were calculated according to eqn (3).
 | (3) |
In this equation, K2 (g mg−1 h−1) is the pseudo-second-order rate constant.
As shown in Fig. 5A, TCP adsorption increased rapidly with time and reached a constant value in 10 h. The kinetic parameters and correlation coefficient (R2) obtained for the plots are given in Table 1. The R2 value of the pseudo-second-order kinetic model was determined to be 0.994 and the calculated qe,cal was found to be very similar to the experimental qe,exp, demonstrating a good fit between the kinetics data and the pseudo-second-order kinetic model.
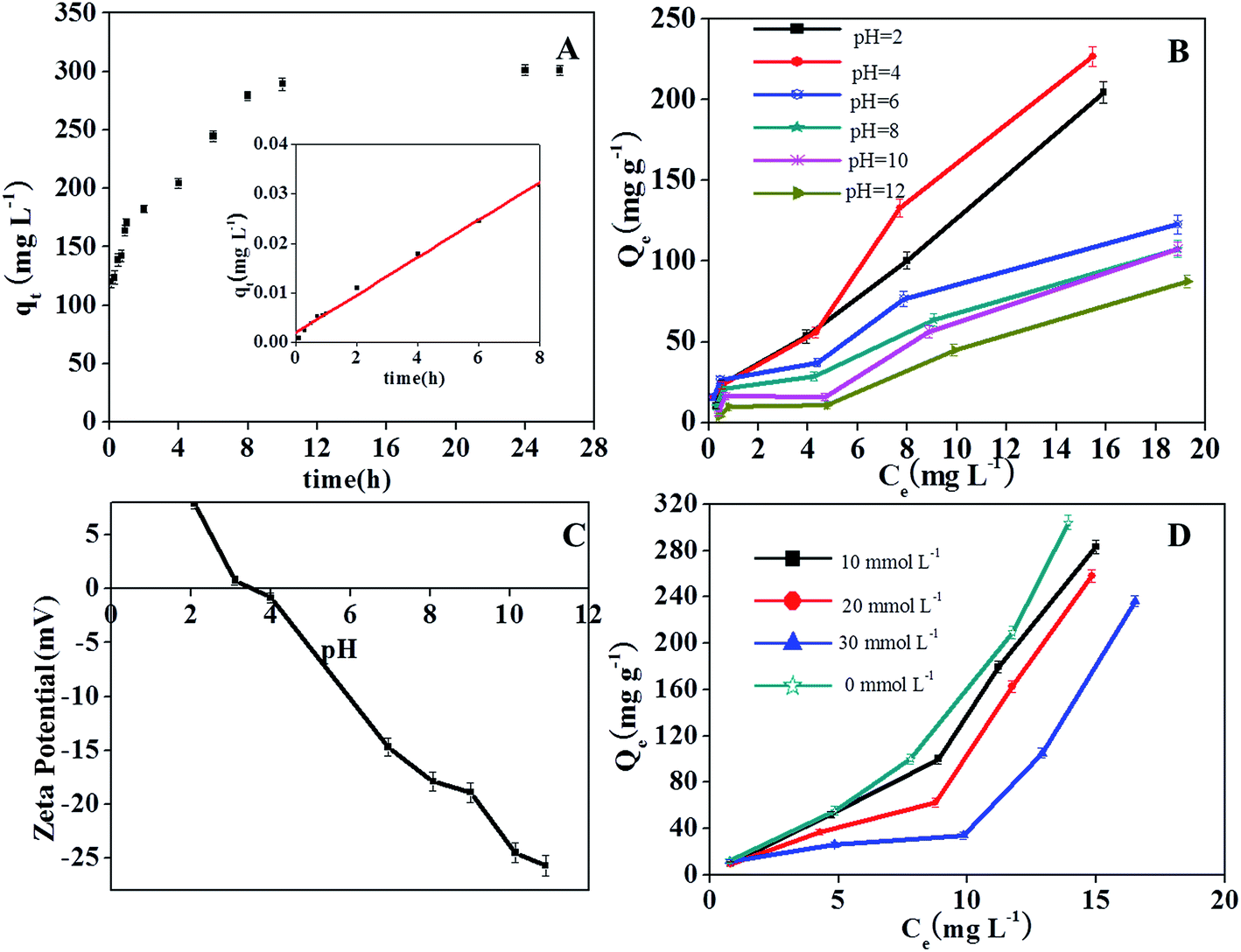 | ||
| Fig. 5 Effect of reaction time (A), solution pH (B) and ionic strength (D) on TCP adsorption onto ZrO2–C; ζ-potential of ZrO2–C at different solution pH values (C). | ||
| qe,exp (mg g−1) | Pseudo-first-order | Pseudo-second-order | |||||
|---|---|---|---|---|---|---|---|
| qe,cal (mg g−1) | k1 (h−1) | R2 | qe,cal (mg g−1) | K2 (h−1) | R2 | ||
| TCP | 306 | 158 | 0.2524 | 0.9146 | 312.5 | 0.0039 | 0.9974 |
3.3 Effect of pH
Solution pH is one of the main factors affecting the adsorption capacity of compounds that can be ionized. The surface chemistry of carbon adsorbents may change with solution pH. These effects may cause the efficiency at which TCP is removed to significantly change as the pH is changed. The uptake of TCP by ZrO2–C was observed to be better in acid solution than in neutral and alkali solutions (Fig. 5B), as has been observed for other carbonaceous materials.37,40,41 In the current study, the capacity of ZrO2–C to adsorb TCP was observed to be greatest at pH 4.0, and then decline as the solution pH was increased. Since the pKa of TCP is pH 5.99,42 the majority of TCP exists in the neutral and un-ionized forms in acid solution, and ionizes gradually and becomes negatively charged at solution pH values greater than 5.99. To interpret the effect of solution pH on TCP adsorption, we also measured ζ-potentials of ZrO2–C in aqueous solutions. The isoelectric point (IEP) of ZrO2–C was found to be about pH 3.5 (Fig. 5C). At pH 3.5–4.0, both TCP and the ZrO2–C surface are thus neutral, which favors hydrophobic and/or π–π stacking interactions between TCP and ZrO2–C. When the solution pH is lower than 3.5, the surface of the material is positively charged and the hydrophobic interaction between TCP and ZrO2–C is weakened. So the adsorption capacity declines in strongly acidic (pH < 3.5) conditions. On the other hand, the neutral form of TCP can also make H-bonds with the oxygen-containing groups of the carbon phase and of the ZrO2 nanoparticles of the adsorbents.In neutral and alkaline solutions, electrostatic repulsion between the negative surface of ZrO2–C and the anionic TCP is expected, and such repulsion would help explain the observed decreased adsorption of TCP on ZrO2–C in these pH conditions. However, the decrease of TCP sorption in the pH range 6–10 was observed to be fairly gradual, and the sorption capacity was still considerable at pH 12. This trend is obviously different than that obtained on most carbon materials such as graphene and graphene oxide,6 multi-walled carbon nanotubes,7 and surfactant-modified bentonite.6 It was reported that the phenolate anions can complex with metal ions on the metal oxides and zeolite surface, and that this complexation proceeds via a kind of charge transfer from phenolate anions to empty d-orbitals of the metal (e.g., Si, Al, Fe, Ti, Mn) on the surface of the metal oxides.11–13,21–23 Okolo et al. suggested that the benzene ring (in particular its π electrons) rather than the hydroxyl substituent of phenols interacts with the synthetic zeolite surface.23 Karunakaran and co-workers43 assumed that the acidic sites on the surface of ZrO2 may coordinate the phenolic oxygen, and/or the basic O− group may be involved in hydrogen bonding with the –OH group of phenol. Since the electron density of phenol rings increases with solution pH, we believe that the considerable adsorption of TCP on ZrO2–C composites at higher solution pH results from complexation of TCP anions with Zr ions on the surfaces of many inlaid ZrO2 NPs. On the other hand, in our previous work, we used magnetic mesoporous carbon, which is composed of graphitic carbon and Fe3C/α-Fe, as an adsorbent to remove TCP as well. The specific surface area and carbon content of this material (220 m2 g−1 and 54.5%, respectively) were observed to be larger than those of ZrO2–C synthesized in this study. However, the adsorption capacity of the magnetic mesoporous carbon to TCP at 303 K was much lower than that achieved on ZrO2–C at pH 4. We deduce that the adsorption of TCP onto ZrO2–C proceeds via a hydrophobic interaction and/or π–π stacking interaction with the carbon phase, by hydrogen bonding with functional groups of ZrO2–C, and/or by complexation with Zr(IV) cations (especially at high solution pH).
3.4 Adsorption isotherm
The effect of solution temperature on TCP adsorption was investigated by carrying out the experiments at 303, 313 and 323 K, respectively. The capacity of ZrO2–C to adsorb TCP was slightly enhanced as the temperature was increased, indicating the endothermic nature of the sorption process. Langmuir and Freundlich isotherm models were used to study the relationship between the level of adsorption of TCP onto ZrO2–C and its equilibrium concentration in aqueous solution. The Langmuir model is widely used for studies of adsorption and assumes that the adsorption takes place on a specific homogeneous surface; in contrast, the Freundlich model assumes that the binding takes place on a heterogeneous adsorption surface with multilayer adsorption.The linear form of the Langmuir isotherm equation is expressed as
 | (4) |
The logarithmic form of the Freundlich equation is
 | (5) |
Table 2 lists the constants and correlation coefficients determined from the two isotherm models for the adsorption of TCP on ZrO2–C. The Freundlich model yielded a better fit with higher R2 values (greater than 0.986), indicating that the Freundlich isotherm is more suitable than the Langmuir model in describing the adsorption of TCP on ZrO2–C. The 1/n value from the Freundlich model was calculated to be below 1, indicating that adsorption of TCP on ZrO2–C is favorable. This result is consistent with TCP sorption on other carbon materials such as loosestrife-based activated carbon,8 activated clay12 and commercial-grade coconut shell-based activated carbon.4
| Solution temperature (K) | Langmuir isotherm model | Freundlich isotherm model | ||||
|---|---|---|---|---|---|---|
| Q0 (mg g−1) | KL (L mg−1) | R2 | K (mg g−1 (L mg−1)1/n) | 1/n | R2 | |
| 303 | 454.5 | 0.090 | 0.7489 | 32.422 | 0.749 | 0.9863 |
| 313 | 476.2 | 0.095 | 0.8139 | 34.655 | 0.743 | 0.9906 |
| 323 | 476.2 | 0.117 | 0.9401 | 38.303 | 0.748 | 0.9941 |
Because of the poor fit between TCP sorption on ZrO2–C and the Langmuir equation, we made a coarse estimate of the ability of ZrO2–C to adsorb organic pollutants by comparing the calculated adsorption capacity of a specific concentration of 20 mg L−1with those of other adsorbents reported in literature. The results are listed in Table 3. Despite the relatively low specific surface areas and small porous volumes, the sorption ability of ZrO2–C for TCP was found to be much higher than those reported previously for many kinds of carbon-based materials. This increased sorption may have resulted from the combined contributions of ZrO2 NPs and the carbon phase of ZrO2–C adsorbents to TCP sorption.
| Adsorbent | T (K) | qe (mg g−1) (C0 = 20 mg L−1) | Reference |
|---|---|---|---|
| Zirconium dioxide–carbon | 303 | 306 | This study |
| Graphene oxide | 303 | 190 | 6 |
| Coconut husk-based activated carbon | 303 | 122 | 5 |
| Graphene | 298 | 50 | 7 |
| CFAC | 293 | 192 | 9 |
| Coconut shell-based activated carbon | 303 | 112 | 4 |
| MMC | 303 | 210 | 10 |
| Commercial activated carbon | 303 | 20 | 4 |
| Activated clay | 303 | 123 | 12 |
3.5 Adsorption thermodynamics
The thermodynamic equations used are as follows:
 | (6) |
| ΔG0 = ΔH0 − TΔS0 | (7) |
In eqn (6), Kd is the distribution coefficient of the adsorbent, equal to qe/Ce, R (8.314 J mol−1 K−1) is the universal gas constant, and T (K) is the temperature. ΔG0 can be derived from ΔH0 and ΔS0.
The values of the thermodynamic parameters obtained for the adsorption of TCP on mesoporous carbon materials are listed in Table 4. The value of ΔH0 was found to be +8.7 kJ mol−1, with the positive value suggesting that the reaction is endothermic, which is consistent with the result that TCP uptake increases with temperature. Also, the main interaction between TCP and the mesoporous carbon materials appears to be of a physical nature since a physical adsorption process is generally indicated by a ΔH0 value less than 40 kJ mol−1 while chemisorption is suggested by a value greater than 40 kJ mol−1. The positive value of ΔS0 suggests that the adsorption is irreversible. The negative value of ΔG0 shows the spontaneous nature of the adsorption process.
| ΔH0 (kJ mol−1) | ΔS0 (J mol−1 K−1) | ΔG0 (kJ mol−1 K−1) | ||
|---|---|---|---|---|
| 303 K | 313 K | 323 K | ||
| 4.105 | 38.49 | −7.56 | −7.94 | −8.33 |
3.6 Effect of ionic strength
The effect of ionic strength on TCP adsorption was investigated by conducting adsorption equilibrium experiments with different concentrations of NaCl at pH 4. As shown in Fig. 5D, the capacity of ZrO2–C to adsorb TCP was found to decrease by 7, 15 and 23% when NaCl concentrations of 10, 20, and 30 mM, respectively, were introduced. This result indicates that hydrophobic and/or π–π stacking interactions between TCP and ZrO2–C are more dominant than non-specific electrostatic interactions, since only the electrostatic interactions can be weakened by increased ionic strength of the solution.4. Conclusion
We have synthesized chrysanthemum-shaped ZrO2–C composites with a high yield by using a simple method. These composites exhibited excellent water stability and a remarkable capacity to adsorb chlorophenols. Hydrophobic and/or π–π stacking interactions and hydrogen bonds between TCP and ZrO2–C contribute to the high level of adsorption of TCP by the ZrO2–C material. Moreover, ZrO2 nanoparticles embedded in the carbon phase can form metal–anion bonds with TCP, which is responsible for the reasonable ability of ZrO2–C to adsorb TCP even in weak alkali solutions. The adsorption of TCP onto ZrO2–C is slightly influenced by temperature and ionic strength. These results support the effectiveness of the method used and provide a reference for the removal of TCP from wastewater.Acknowledgements
This work was jointly supported by National Basic Research Program of China (2015CB93203), National High Technology Research and Development Program of China (2013AA065201), Strategic Priority Research Program of the Chinese Academy of Sciences (XDB14010201), State Environmental Protection Welfare Scientific Research Project (201409037), the National Natural Science Foundation of China (21537004, 21277152, 21277002, 41222026), and the Open Foundation of State Key Laboratory of Environmental Criteria and Risk Assessment, Chinese Research Academy of Environmental Sciences (No. SKLECRA2013OFP16).References
- R. S. Ojha Priyanka, S. Kunwar and S. Afr, J. Chem. Eng., 2014, 19, 1–21 Search PubMed.
- D. Krishnaiah, S. M. Anisuzzaman, A. Bono and R. Sarbatly, J. King Saud Univ., Sci., 2013, 25, 251–255 CrossRef PubMed.
- S. Wang, H. Niu, T. Zeng, X. Ma, Y. Cai and X. Zhao, CrystEngComm, 2014, 16, 5598 RSC.
- M. Radhika and K. Palanivelu, J. Hazard. Mater., 2006, 138, 116–124 CrossRef CAS PubMed.
- B. H. Hameed, I. A. W. Tan and A. L. Ahmad, Chem. Eng. J., 2008, 144, 235–244 CrossRef CAS PubMed.
- Z. Pei, L. Li, L. Sun, S. Zhang, X.-q. Shan, S. Yang and B. Wen, Carbon, 2013, 51, 156–163 CrossRef CAS PubMed.
- G. C. Chen, X. Q. Shan, Y. S. Wang, B. Wen, Z. G. Pei, Y. N. Xie, T. Liu and J. J. Pignatello, Water Res., 2009, 43, 2409–2418 CrossRef CAS PubMed.
- J. Fan, J. Zhang, C. Zhang, L. Ren and Q. Shi, Desalination, 2011, 267, 139–146 CrossRef CAS PubMed.
- C. Namasivayam and D. Kavitha, J. Environ. Eng. Sci., 2004, 46, 217–232 CAS.
- S. Liu, S. Li, H. Niu, T. Zeng, Y. Cai, C. Shi, B. Zhou, F. Wu and X. Zhao, Microporous Mesoporous Mater., 2014, 200, 151–158 CrossRef CAS PubMed.
- R. I. Yousef and B. El-Eswed, Colloids Surf., A, 2009, 334, 92–99 CrossRef CAS PubMed.
- B. H. Hameed, Colloids Surf., A, 2007, 307, 45–52 CrossRef CAS PubMed.
- Y. Zhang, R. G. Mancke, M. Sabelfeld and S. U. Geissen, J. Hazard. Mater., 2014, 271, 178–184 CrossRef CAS PubMed.
- T. S. Anirudhan and M. Ramachandran, J. Water Process Eng., 2014, 1, 46–53 CrossRef PubMed.
- J. A. Mielczarski, J. Bandara and J. Kiwi, Appl. Catal., B, 2001, 34, 321–333 CrossRef.
- H. Peng, J. Cui, H. Zhan and X. Zhang, Chem. Eng. J., 2015, 264, 316–321 CrossRef CAS PubMed.
- M. Pera-Titus, V. Garcıa-Molina, M. A. Baños, J. Giménez and S. Esplugas, Appl. Catal., B, 2004, 47, 219–256 CrossRef CAS PubMed.
- H. Wang and J. L. Wang, J. Hazard. Mater., 2008, 154, 44–50 CrossRef CAS PubMed.
- M. S. Miao, Y. J. Zhang, L. Shu, J. Zhang, Q. Kong and N. Li, Int. Biodeterior. Biodegrad., 2014, 95, 61–66 CrossRef CAS PubMed.
- A. G. Jesus, F. J. Romano-Baez, L. Leyva-Amezcua, C. Juarez-Ramirez, N. Ruiz-Ordaz and J. Galindez-Mayer, J. Hazard. Mater., 2009, 161, 1140–1149 CrossRef CAS PubMed.
- K. H. Kung and M. B. McBride, Environ. Toxicol. Chem., 1991, 10, 441–448 CrossRef PubMed.
- M. B. McBride and K. H. Kung, Environ. Sci. Technol., 1991, 25, 702–709 CrossRef.
- B. Okolo, C. Park and M. A. Keane, J. Colloid Interface Sci., 2000, 226, 308–317 CrossRef CAS.
- M. Ó. C. M. J. Hynes, J. Inorg. Biochem., 2001, 85, 131–142 CrossRef CAS.
- K. F. Pirker, M. C. Baratto, R. Basosi and B. A. Goodman, J. Inorg. Biochem., 2012, 112, 10–16 CrossRef CAS PubMed.
- M. J. Hynes and M. Ó'Coinceanainn, J. Inorg. Biochem., 2001, 84, 1–12 CrossRef.
- X. Zhang, Y. Hou, P. Hu and C. Hong, J. Eur. Ceram. Soc., 2012, 32, 3463–3468 CrossRef CAS PubMed.
- S. Tascioglu, O. Sendil and S. Beyreli, Anal. Chim. Acta, 2007, 590, 217–223 CrossRef CAS PubMed.
- B. I. Ipe, S. Cherumuttathu, H. Y. Karuvath and T. K. George, J. Phys. Chem., 2007, 111, 12839–12847 CrossRef PubMed.
- D. Feng, T. F. Liu, J. Su, M. Bosch, Z. Wei, W. Wan, D. Yuan, Y. P. Chen, X. Wang, K. Wang, X. Lian, Z. Y. Gu, J. Park, X. Zou and H. C. Zhou, Nat. Commun., 2015, 6, 5979 CrossRef PubMed.
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC.
- H. R. Abid, H. M. Ang and S. Wang, Nanoscale, 2012, 4, 3089–3094 RSC.
- J. David, G. Trolliard, C. Volkringer, T. Loiseau and A. Maître, Phys. Rev. Lett., 2015, 5, 51650–51661 CAS.
- A. M. Ebrahim and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2013, 5, 10565–10573 CAS.
- F. Heshmatpour and R. B. Aghakhanpour, Adv. Powder Technol., 2012, 23, 80–87 CrossRef CAS PubMed.
- A. K. Singh, Adv. Powder Technol., 2010, 21, 609–613 CrossRef CAS PubMed.
- J. B. DeCoste, G. W. Peterson, H. Jasuja, T. G. Glover, Y. G. Huang and K. S. Walton, J. Mater. Chem., 2013, 1, 5642 RSC.
- J. Long, S. Wang, Z. Ding, S. Wang, Y. Zhou, L. Huang and X. Wang, Chem. Commun., 2012, 48, 11656–11658 RSC.
- J. Wang, T. Shi and X. Jiang, Nanoscale Res. Lett., 2008, 4, 240–246 CrossRef CAS PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- Y. Tanaka, M. Nakai, T. Akahori, M. Niinomi, Y. Tsutsumi, H. Doi and T. Hanawa, Corros. Sci., 2008, 50, 2111–2116 CrossRef CAS PubMed.
- I. Sabbah and M. Rebhun, Water Environ. Res., 1997, 69, 1032–1038 CrossRef CAS.
- C. Karunakaran, R. Dhanalakshmi and P. Gomathisankar, Spectrochim. Acta, Part A, 2012, 92, 201–206 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |