
Fabrication and properties of low crystallinity nanofibrillar cellulose and a nanofibrillar cellulose–graphene oxide composite
 ab
and
ab
and
Abstract
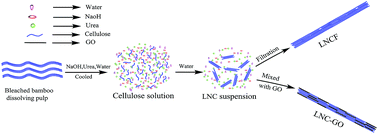
Nanofibrillar cellulose with low crystallinity (LNC) was fabricated by adding water into a cellulose solution, which was facile, efficient and environmentally friendly. A strong and translucent nanofibrillar cellulose film with low crystallinity (LNCF) was prepared from a LNC suspension by filtration. Transmission electron microscopy showed that LNC was individually separated whiskers with a length of 200 nm, a width of 20 nm. Structure characterization of LNC showed that the main crystal form of LNC was cellulose II and the crystallinity was as low as 22.3%. LNCF exhibited excellent optical transparency of 91.7% when the thickness of the film was 40 μm. The tensile strength and young's modulus of LNCF reached 64.7 MPa and 3062.8 MPa respectively. Meanwhile, a nanofibrillar cellulose–graphene oxide composite (LNC–GO) with a layer by layer structure was obtained by mixing LNC with graphene oxide at the mass ratio of 1 : 1, and the tensile strength and young's modulus of LNC–GO were 84.3 MPa and 4406.2 MPa respectively.
 Please wait while we load your content...
Please wait while we load your content...

