
MOLPRINT 2D-based identification and synthesis of novel chromene based small molecules that target PLA2: validation through chemo- and bioinformatics approaches†
Abstract
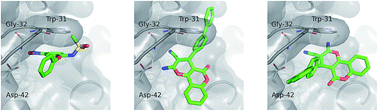
Phospholipase A2 (PLA2) is known to regulate inflammation and hence it is considered as a validated drug-target by medicinal chemists. In this report, we have identified and considered a highly ranked ligand from the ZINC-drug-like compounds database that targets PLA2 via the MOLPRINT-2D based chemoinformatics drug-design approach. The computationally predicted lead molecule was found to contain a core moiety of a chromene ring, which is well known for its varied biological properties. Here, a novel and efficient retro-synthetic protocol for the synthesis of highly substituted chromene libraries was made. A one-pot synthesis of chromene was carried out using different aromatic primary alcohols, malononitrile and 4-hydroxy coumarin in the presence of a mild oxidant mixture called T3P®–DMSO, followed by a Suzuki coupling reaction to obtain the lead molecules. All of the tested compounds of the chromene series displayed inhibition of the venom PLA2 in the range of 12 to 68 μM. Among the tested compounds, 2-amino-4-(2′-methyl-[1,1′-biphenyl]-4-yl)-5-oxo-4,5-dihydropyrano[3,2-c]chromene-3-carbonitrile (7b) showed maximum inhibitory efficacy against venom PLA2 with an IC50 value of 12.5 μM. Furthermore, the designed PLA2 ligands bound to the active site of venom PLA2, whose binding affinity was comparable to nimesulide, indicating that the chromene moiety containing ligands could be novel lead-structures that serve as anti-inflammatory agents.
 Please wait while we load your content...
Please wait while we load your content...

