
Design, synthesis and biological evaluation of novel unsymmetrical azines as quorum sensing inhibitors†
Abstract
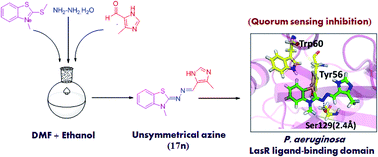
Targeting quorum sensing signals using quorum sensing inhibitors has opened new avenues for the application of known antibiotics. In this context, twenty five unsymmetrical azines were synthesised and evaluated as quorum sensing inhibitors. An efficient one-pot procedure was adopted that directly links 3-methyl-2-(methylthio)benzo[d]thiazol-3-ium salt, hydrazine hydrate and substituted aldehyde to give the designed compounds. The synthesized compounds were preliminarily tested for their potential to inhibit CviR receptor based QS signals in Chromobacterium violaceum. The bioassay screening results suggested that two compounds exhibited potent QS inhibition activity against CviR receptor, showing violacein inhibition (>50%) at 200 μM. Further, the putative positive hits were checked for their potential to inhibit LasR receptor-based QS using the PlasB-gfp(ASV) biomonitor strain of Pseudomonas aeruginosa. These compounds were found to inhibit the QS-mediated GFP signals in a dose dependant manner. Two active compounds also exhibited biofilm clearance at 50 μM concentration. Docking studies were performed to examine their potential to bind to the LasR protein of Pseudomonas aeruginosa.
 Please wait while we load your content...
Please wait while we load your content...

