
Facile synthesis of submillimeter sized graphene oxide by a novel method†
Tao Xua,
Qinghan Meng*a,
Tianze Shenb and
Bing Cao*b
aThe Key Laboratory of Beijing City on Preparation and Processing of Novel Polymer Materials, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: qhmeng@mail.buct.edu.cn
bCollege of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: bcao@mail.buct.edu.cn
First published on 24th August 2015
Abstract
High-quality graphene oxide (GO) was synthesized in two steps. First, graphite on the anode was expanded in an aqueous sulfuric acid solution at a constant direct-current power supply, then it is used to synthesize GO by an improved Hummers method. The synthesized GO had a low layer number (less than five layers) and large lateral size (at least 300 μm).
Graphene (GN), a two-dimensional honeycomb sp2 carbon lattice, exhibits superior electrical conductivity and a high charge carrier mobility because electron tunnelling occurs within its structure allowing electron movement at relativistic speeds.1,2 GN is used in a wide range of devices such as organic photovoltaics,3–6 high speed transistors,7,8 field emission electrodes,9 transparent conducting films,10 lithium ion batteries,11–13 hydrogen storage14 and supercapacitors.15 GO, which is an oxygen-rich carbonaceous material, contains both sp2 and sp3 hybridized carbon atoms. The sp3 hybridized carbon atoms in GO are covalently bonded with oxygen-containing functional groups such as carboxyl, carbonyl, epoxy groups, hydroxyl, etc.16 The covalent oxygen-containing functional groups in GO bring remarkable modification of the graphene (GN) electronic structure. Therefore, the chemical composition of GO, which can be electrochemically engineered, allows tunability of its optoelectronic properties.17,18 In recent years, GO has also been directly used for electronics.3,4,10,19 Because of rigorous requirements of specific surface area, optical transparency, electrical conductivity and resistance for GN at electronics industry, GN is usually produced using chemical vapor deposition (CVD) method in order to control the quality of the carbon lattice.20 But CVD method also exhibits several disadvantages, including low productivity, non-viability for commercialization, time-consuming nature, and use of high temperature. These characteristics are unsuitable for large-scale production of GN. Chemical graphite to GO exfoliation and GO to GN conversion by chemical or thermal reduction have received considerable attention because these methods exhibit low-cost and mass production capability. The GO products, synthesized by chemical exfoliation,21–25 have a common characteristic that the lateral dimensions of GO flakes are small, just ranged from several hundreds of nanometers to several dozens of micrometers. The small lateral dimensions of GO severely restricted its application in electronic industry.
In this study, hundreds of micrometers sized GO was synthesized in two steps. First, the expanded graphite flakes with particle size larger than 300 μm were synthesized by electrochemical intercalation. Graphite sheets were used as electrodes. The distance between cathode and anode was maintained at 2 cm, and the concentration of the aqueous H2SO4 solution was 2 mol L−1. A constant voltage of 5 V from the DC power supply was applied between the two electrodes for 5 min. This step aided graphite electrode on the anode to become wet and caused gentle intercalation of SO42− to the grain boundary of graphite, which expanded the structures. The expanded graphite sheets were washed with deionized water to remove residual electrolytes on the surface of sheets, and then dried at 80 °C under atmospheric pressure for 8 h. The dried graphite sheets were ground using a grinder to produce flakes, and the flakes were sonicated in anhydrous ethanol for 5 minutes. Thereafter, the flakes were passed through a sieve with a pore diameter of 300 μm and dried at 60 °C under atmospheric pressure for 1 h. The expanded graphite flakes with particle size larger than 300 μm were selected to prepare GO. Second, high-quality GO was synthesized using the expanded graphite flakes by an improved Hummers method. Around 70 mL of concentrated H2SO4 was added to 4.0 g of the mixture of expanded graphite flakes under stirring in an ice-water bath. Exactly 9.0 g of KMnO4 was slowly added to the mixture so that the reaction temperature was increased by no more than 10 °C. The reaction was maintained for 30 min, and the mixture was transferred to a water bath at 40 °C and continuously stirred for 30 min. After adding 150 mL of deionized water, the mixture was heated and maintained at 95 °C for 15 min. After heating, 1200 mL of deionized water and 5 mL of 30% H2O2 were sequentially added to the mixture, turning its color from ash brown to golden yellow. The mixture was then filtered through a 0.22 μm (pore size) membrane and washed with 300 mL of diluted HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) followed by deionized water until the pH became 7. The obtained graphite oxide was dispersed in water and sonicated for 30 min to produce exfoliated GO, which was centrifuged at 4000 rpm for 15 min to remove unexfoliated graphite. GO was vacuum freeze dried, and the final product was collected for further characterization.
:10) followed by deionized water until the pH became 7. The obtained graphite oxide was dispersed in water and sonicated for 30 min to produce exfoliated GO, which was centrifuged at 4000 rpm for 15 min to remove unexfoliated graphite. GO was vacuum freeze dried, and the final product was collected for further characterization.
Fig. 1a is the schematic representation of the pre-oxidation mechanism. When a constant voltage from the DC power supply was applied between the two graphite electrodes, the initial low voltage helps to wet the sample (Fig. 1b) and causes gentle intercalation of O2 (released by side reactions) and SO42− ions to the grain boundary of graphite.26 This behavior destroyed the van der Waals force between the graphite interlayer, and expanded the structures of graphite precursor with O2, SO42− ions and H2O (Fig. 1c). Fig. 1d shows the digital images of the expanded graphite. The thickness of the expanded portion (d′ = 1.7 nm) is three times of the unexpanded portion (d = 0.6 nm). After the four steps of water washing, atmospheric drying, grinding and sieving, the expanded graphite flakes with particle size larger than 300 μm (Fig. 1e) were used to prepare GO using improved Hummers method. As described on the scheme (Fig. 1e and f), the combination H2SO4/KMnO4 used in Hummers method acted as a chemical “scissor” that further destroyed the van der Waals force between graphite layers and facilitated chemical oxidation exfoliation, forming graphite oxide in which every single-layer GO is sandwiched by the layers of oxygen-containing functional groups.21,27,28
 | ||
| Fig. 1 The schematic representation of preparation process and mechanism for GO formation from graphite precursor. | ||
In the pre-oxidation process of graphite sheets, we selected the expanded graphite flakes with particle size larger than 300 μm by means of crushing and screening. The artificial screening of pre-oxidated graphite precursor particles greatly restrained the reduction of the crystallite sizes of the GO caused by rupture of the graphitic sheets in an electrochemical intercalation process. Meanwhile, the pre-oxidation process destroyed the van der Waals force between the graphite interlayer and increased the spacing between graphite layers. This behavior had two distinct advantages for followed synthesis of GO by using improved Hummers method. Firstly, the increase of the layer spacing between graphite layers was in favor of the further intercalation of oxidants. Secondly, the destruction of the van der Waals force between the graphite interlayers decreased the difficulty to prepare GO by followed improved Hummers method, and restrained excessive basal plane oxidations caused enormous strain in the graphitic sheets which resulted rupture of the graphitic sheets along the a-axis (axis in the carbon layer planes) as well.29
The detailed morphology and crystalline structure of the expanded graphite flakes after the electrochemical pre-oxidation process were investigated by SEM, TEM, HR-TEM and AFM. The lateral dimensions of expanded graphite flakes reached 300 μm according to their TEM (Fig. 2a) and SEM (Fig. 2b) images. TEM images revealed that the transparency of expanded graphite flakes were poor, which inferred expanded graphite flakes had a great number of layers. It was obvious that expanded graphite flakes had plenty of layers according to HR-TEM (Fig. 2c) image, too. AFM was performed to investigate the topographic height of GO. Because of the test range limitation of AFM (30 μm × 30 μm), expanded graphite flakes was sonicated for 1 h to be broken into microsized flakes before AFM test. AFM (Fig. 2d) shows that the average thickness of expanded graphite flakes was 407 nm. The observed thickness indicated that multilayer expanded graphite flakes formed, which confirmed the earlier conclusion drawn based on Fig. 2a and c.
 | ||
| Fig. 2 (a) TEM, (b) SEM, (c) HR-TEM and (d) AFM of expanded graphite flakes after the electrochemical pre-oxidation process. | ||
The XRD (Fig. 3a) pattern of expanded graphite flakes gives a (002) reflection peak at 2θ = 26.4°, with an interlayer distance of 0.34 nm. Meanwhile, the XRD pattern of expanded graphite flakes displayed a (001) reflection peak at 2θ = 13.8°, which corresponded to a d space of 0.64 nm. The large interlayer spacing of GO sheets were attributed to a handful of oxygenated functional groups and a large amount of H2O and SO42− ions in expanded graphite flakes layers. Both the (002) diffraction line observed in graphite and (001) reflection line in GO were present in the XRD diffraction spectrum of expanded graphite flakes. Thus, expanded graphite flakes was uncompletely oxidized to GO after the electrochemical pre-oxidation process.30 Raman spectroscopic (Fig. 3b) shows a signal of the pristine sp2 GN lattice at 1582 cm−1, which corresponded to the G band, whereas sp3 defects in the sp2 lattice produced a prominent signal at 1353 cm−1. The ID/IG ratio of expanded graphite flakes was 0.21, which suggested that expanded graphite flakes displayed a low degree of oxidation, and a small quantity of sp2 lattice was destroyed and became disordered sp3 lattice.31 Fig. 3c shows the FT-IR spectrum of expanded graphite flakes. A sharp and broad intense band was observed at 3420 cm−1, corresponding to –OH stretching vibrations. These stretching vibrations were due to the electrochemical intercalation of graphite caused by using H2SO4 solution as an electrolyte. The appearance of sharp peaks at 1629, 1230, and 1069 cm−1 in the expanded graphite flakes were attributed to COOH, C–OH, and C–O–C stretching vibrations, respectively. The presence of the peak at 850 cm−1 was attributed to aromatic C–H stretching and out-of-plane bending vibration. FT-IR spectrum (Fig. 3c) proves the presence of free (nonbonded) SO42− (at 1009 cm−1).32 Fig. 3d shows the details of the C1s signal of expanded graphite flakes. The spectrum showed four types of C bonds, namely, C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 284.5 eV, C–O at 286.5 eV and O–CO at 288.9 eV. The atomic percentage, measured by XPS, were C (71.47%), O (24.74%) and S (3.79%). Deduction of the O atom (4 times of S atomic percentage) derived from SO42− ions, the atomic percentage of O, which originated from oxygen-containing functional groups, was 11.82%. Therefore, the C/O atomic ratios of expanded graphite flakes was 5.9. Results showed that expanded graphite flakes was a low degree of oxidation during the electrode–electrolyte interaction process.
C at 284.5 eV, C–O at 286.5 eV and O–CO at 288.9 eV. The atomic percentage, measured by XPS, were C (71.47%), O (24.74%) and S (3.79%). Deduction of the O atom (4 times of S atomic percentage) derived from SO42− ions, the atomic percentage of O, which originated from oxygen-containing functional groups, was 11.82%. Therefore, the C/O atomic ratios of expanded graphite flakes was 5.9. Results showed that expanded graphite flakes was a low degree of oxidation during the electrode–electrolyte interaction process.
 | ||
| Fig. 3 (a) XRD, (b) Raman, (c) FT-IR and (d) XPS of expanded graphite flakes after the electrochemical pre-oxidation process. | ||
Fig. 4a shows the TEM image of GO. TEM images revealed that the lateral dimensions of GO flakes reached 300 μm. Fig. 4a displayed that GO flakes were rippled and entangled with one another, and were transparent and extremely stable under an electron beam. The most transparent and featureless regions indicated by the arrows in Fig. 4a were probably the monolayer of GO. Fig. 4b shows the SEM images of GO. GO flakes displayed wrinkled or folded morphology. SEM images revealed that the lateral dimensions of GO flakes reached at least 300 μm, which was range 10 to 100 times larger than previously reported values.21–23,33 The HR-TEM image (Fig. 4c) indicated that a single layer of GO formed, which confirmed our earlier conclusion. This result was estimated based on statistical sampling of exfoliated GO flakes, i.e., by calculating the layers of 50 GO flakes in the HR-TEM image (Fig. 4d). Approximately 36% of GO flakes were monolayer, nearly 54% exhibited two to five layers, and nearly 10% had six to ten layers.
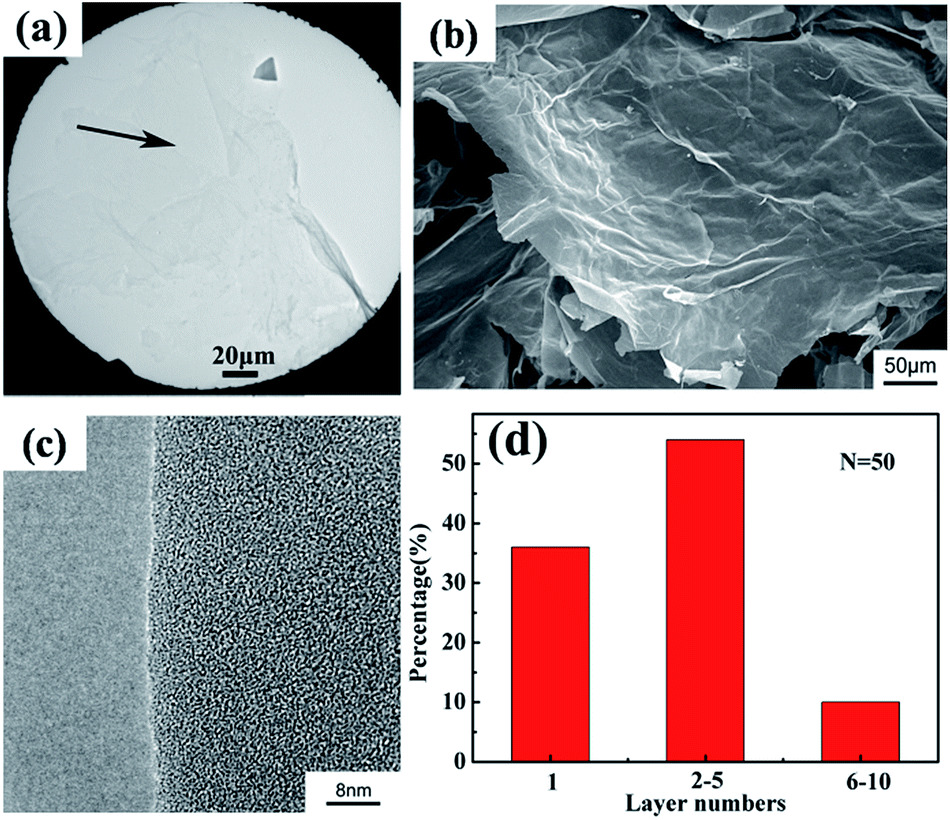 | ||
| Fig. 4 (a) TEM, (b) SEM and (c) HR-TEM of GO; (d) layer numbers distribution histogram for exfoliated GO sheets, as estimated from the corresponding HR-TEM analysis. | ||
AFM was performed to investigate the topographic height of GO. Because of the test range limitation of AFM (30 μm × 30 μm), the GO was sonicated for 1 h to be broken into microsized GO before AFM test. Fig. 5a shows that the average thickness of GO was 1 nm. The observed thickness indicated that a single layer of GO formed,34 which confirmed the earlier conclusion drawn based on Fig. 4. Fig. 5b shows the XRD results of graphite and GO. A difference was found between the spacing between layers, with an interlayer distance of 0.34 nm for pure graphite, as indicated by the (002) reflection peak near 2θ = 26.4°. Meanwhile, the XRD pattern of GO displayed a (001) reflection peak at 2θ = 11.0°, which corresponded to a d space of 0.80 nm. The large interlayer spacing of GO sheets were attributed to the oxygenated functional groups in GO; these functional groups were introduced when graphite underwent harsh oxidation.21 The (002) diffraction line observed in graphite was not present in the XRD diffraction spectrum of GO. Thus, graphite was completely oxidized to GO.30 Raman spectroscopic (Fig. 5c) shows a signal of the pristine sp2 GN lattice at 1582 cm−1, which corresponded to the G band, whereas sp3 defects in the sp2 lattice produced a prominent signal at 1355 cm−1. The ID/IG ratio of GO was 0.94, which suggested that GO displayed a high degree of oxidation, and a large number of sp2 lattice was destroyed and became disordered sp3 lattice.31 Fig. 5d shows the FT-IR spectrum of GO. A sharp and broad intense band was observed at 3410 cm−1, corresponding to –OH stretching vibrations. These stretching vibrations were due to the electrochemical intercalation of graphite caused by using H2SO4 solution as an electrolyte and the oxidation of graphite by an improved Hummers method. The peaks at 2926 and 2848 cm−1 represented the asymmetric and symmetric vibrations of CH2 groups. The appearance of sharp peaks at 1736, 1623, 1226 and 1053 cm−1 in the GO were attributed to CO, COOH, C–OH and C–O–C stretching vibrations, respectively. The presence of the peak at 853 cm−1 was attributed to aromatic C–H stretching and out-of-plane bending vibration. FT-IR results confirmed the presence of oxygen-containing functional groups, which may increase water dispersibility.35 Fig. 5e shows the details of the C1s signal of GO. The spectrum showed four types of C bonds, namely, C–C/CC at 284.5 eV, C–O at 286.5 eV, CO at 287.7 eV, and O–CO at 288.9 eV. The C/O atomic ratios of GO, which were measured by XPS, was 2.1; this value was close to the low limit of previous results (1.8 to 4.7) for GO products.33,36–38 Peak intensities of intact C (C–C/CC) and oxygenated C atoms in the XPS spectrum were 40.63% and 59.37%, respectively. Results showed that GO was substantially oxidized during the preparation process. This result was due to several factors, namely, electrode–electrolyte interaction, oxidation exfoliation of electrochemical method, and serious oxidation caused by the harsh oxidizers H2SO4 and KMnO4 when GO was prepared by an improved Hummers method.
 | ||
| Fig. 5 (a) AFM, (b) XRD, (c) Raman, (d) FT-IR and (e) XPS of GO. | ||
Comparison with GO prepared by traditional Hummers method, GO synthesized in this study has two obvious advantages. First, GO flakes had large lateral size reached 300 μm. GN with large lateral size means that it has a high specific surface area, excellent mechanical strength, good elasticity, superior thermal conductivity and optical transparency and high electrical conductivity.1,2,39,40 Second, GO displayed a high degree of oxidation (ID/IG = 0.94, C/O = 2.1), this is to say GO possessed abundant of oxygen functional groups. GO with abundant of oxygen functional groups is considered as a promising materials for composite material applications owing to its excellent aqueous processability, amphiphilicity, surface functionalizability, surface enhanced Raman scattering property, and fluorescence quenching ability.3–6,41,42
Conclusions
Submillimeter sized GO was prepared by pre-oxidizing graphite sheets and synthesizing GO using an improved Hummers method. Test results indicated that GO exhibited excellent properties. About 90% of GO flakes had less than five layers, and the lateral size of GO reached 300 μm. Meanwhile, GO displayed a high degree of oxidation, low C/O ratio (2.11), and high oxygenated C content (59.37%). Synthesizing GO with the novel method may have potential use in several practical applications as a precursor that can be fabricated into electronics having controlled compositions and microstructures.Acknowledgements
It is a pleasure to acknowledge the generous financial support of this research by the National Natural Science Foundation of China (51372011).Notes and references
- J. Lee, L. Tao, Y. Hao, R. S. Ruoff and D. Akinwande, Appl. Phys. Lett., 2012 DOI:10.1063/1.3702570.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- E. Stratakis, K. Savva, D. Konios, C. Petridis and E. Kymakis, Nanoscale, 2014, 6, 6925–6931 RSC.
- G. Kakavelakis, D. Konios, E. Stratakis and E. Kymakis, Chem. Mater., 2014, 26, 5988–5993 CrossRef CAS.
- D. Konios, C. Petridis, G. Kakavelakis, M. Sygletou, K. Savva, E. Stratakis and E. Kymakis, Adv. Funct. Mater., 2015, 25, 2213–2221 CrossRef CAS PubMed.
- N. Balis, D. Konios, E. Stratakis and E. Kymakis, Chem. Nanomater., 2015 DOI:10.1002/cnma.201500044.
- F. Schwierz, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- G. Viskadouros, D. Konios, E. Kymakis and E. Stratakis, Appl. Phys. Lett., 2014 DOI:10.1063/1.4902130.
- Q. Zhang, Y. Di, C. M. Huard, L. J. Guo, J. Wei and J. Guo, J. Mater. Chem. C, 2015, 3, 1528–1536 RSC.
- J. Qin, W. Lv, Z. Li, B. Li, F. Kang and Q. H. Yang, Chem. Commun., 2014, 50, 13447–13450 RSC.
- J. Hassoun, F. Bonaccorso, M. Agostini, M. Angelucci, M. G. Betti, R. Cingolani, M. Gemmi, C. Mariani, S. Panero, V. Pellegrini and B. Scrosati, Nano Lett., 2014, 14, 4901–4906 CrossRef CAS PubMed.
- L. Zhan, S. Yang, Y. Wang, Y. Wang, L. Ling and X. Feng, Adv. Mater. Interfaces, 2014 DOI:10.1002/admi.201300149.
- V. Tozzini and V. Pellegrini, Phys. Chem. Chem. Phys., 2013, 15, 80–89 RSC.
- J. Liu, Y. Xue, M. Zhang and L. Dai, MRS Bull., 2012, 37, 1265–1272 CrossRef CAS.
- G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, K. A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS PubMed.
- F. Zhang, H. Cao, D. Yue, J. Zhang and M. Qu, Inorg. Chem., 2012, 51, 9544–9551 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- L. Peng, Z. Xu, Z. Liu, Y. Wei, H. Sun, Z. Li, X. Zhao and C. Gao, Nat. Commun., 2015 DOI:10.1038/ncomms6716.
- L. Sun and B. Fugetsu, Mater. Lett., 2013, 109, 207–210 CrossRef CAS PubMed.
- D. Konios, M. M. Stylianakis, E. Stratakis and E. Kymakis, J. Colloid Interface Sci., 2014, 430, 108–112 CrossRef CAS PubMed.
- C. T. J. Low, F. C. Walsh, M. H. Chakrabarti, M. A. Hashim and M. A. Hussain, Carbon, 2013, 54, 1–21 CrossRef CAS PubMed.
- V. V. Avdeev, L. A. Monyakina, I. V. Nikol'skaya, N. E. Sorokina and K. N. Semenenko, Carbon, 1992, 30, 819–823 CrossRef CAS.
- N. E. Sorokina, M. A. Khaskov, V. V. Avdeev and I. V. Nikol'skaya, Russ. J. Gen. Chem., 2005, 75, 162–168 CrossRef CAS.
- D. Roy Chowdhury, C. Singh and A. Paul, RSC Adv., 2014, 4, 15138–15145 RSC.
- H. L. Poh, F. Sanek, A. Ambrosi, G. Zhao, Z. Sofer and M. Pumera, Nanoscale, 2012, 4, 3515–3522 RSC.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'Homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- C.-Y. Su, A.-Y. Lu, Y. Xu, F.-R. Chen, A. N. Khlobystov and L.-J. Li, ACS Nano, 2011, 5, 2332–2339 CrossRef CAS PubMed.
- Y. Xu, K. Sheng, C. Li and G. Shi, J. Mater. Chem., 2011, 21, 7376–7380 RSC.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- C. Li and G. Shi, Nanoscale, 2012, 4, 5549–5563 RSC.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- W. Chen and L. Yan, Nanoscale, 2010, 2, 559–563 RSC.
- C. Botas, P. Álvarez, P. Blanco, M. Granda, C. Blanco, R. Santamaría, L. J. Romasanta, R. Verdejo, M. A. López-Manchado and R. Menéndez, Carbon, 2013, 65, 156–164 CrossRef CAS PubMed.
- K. S. Novoselov, Z. Jiang, Y. Zhang, S. V. Morozov, H. L. Stormer, U. Zeitler, J. C. Maan, G. S. Boebinger, P. Kim and A. K. Geim, Science, 2007, 315, 1379 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS PubMed.
- C. Chung, Y.-K. Kim, D. Shin, S.-R. Ryoo, B. H. Hong and D.-H. Min, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- J. Li, F. Wang and C.-Y. Liu, J. Colloid Interface Sci., 2012, 382, 13–16 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12901j |
| This journal is © The Royal Society of Chemistry 2015 |