
A sensitive immobilization-free electrochemical assay for T4PNK activity based on exonuclease III-assisted recycling
Abstract
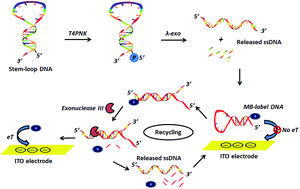
Coupling the properties of T4 polynucleotide kinase (T4PNK) catalyzing the transfer of ATP γ-phosphate residue to 5′-hydroxyl termini of nucleic acids, with lambda exonuclease (λ-exo), a highly processive 5′–3′ exonuclease, digesting the 5′-phosphorylated strand of a double DNA to produce single-strand DNA and mononucleotides, this work develops a highly sensitive immobilization-free electrochemical method for the detection of T4PNK activity based on λ-exo and exonuclease III-assisted signal amplification. Upon the reaction of T4PNK and λ-exo on substrate-DNA, single-stranded DNA segments (released-ssDNA) are dissociated to hybridize with a methylene blue-labeled hairpin probe (MB-DNA) and then the digestion of MB-DNA from the blunt 3 terminus by exonuclease III is activated, resulting in the release of MB-labeled mononucleotides and the complementary DNA segment, followed by the latter hybridizing with another MB-DNA to initiate the cycling process. With a smaller size and less negative charge, the MB-labeled mononucleotide thus diffuses facilely to the negatively charged indium tin oxide (ITO) electrode, actuating an amplified electrochemical signal, and the detection limit of the proposed assay can reach as low as 0.005 U mL−1. Additionally, this assay can avoid the sophisticated probe immobilization processes. Therefore, this strategy exhibits the merits of high sensitivity, simplicity, and being immobilization-free for electrochemical assay of T4PNK activity, which is consequently believed to bear considerable potential as a detection platform for related researches.
 Please wait while we load your content...
Please wait while we load your content...

