
Selective preconcentration and online collection of charged molecules using ion concentration polarization†
Jihye Choi‡
a,
Keon Huh‡a,
Dustin Jaesuk Moona,
Hyomin Leea,
Seok Young Sona,
Kihong Kima,
Hee Chan Kimb,
Jong-Hee Chaec,
Gun Yong Sungd,
Ho-Young Kimef,
Jong Wook Hong*g and
Sung Jae Kim*afh
aDepartment of Electrical and Computer Engineering, Seoul National University, Seoul 151-744, South Korea. E-mail: gates@snu.ac.kr; Tel: +82-2-880-1665
bDepartment of Biomedical Engineering, Seoul National University, Seoul 110-799, South Korea
cDepartment of Pediatrics, Seoul National University, Seoul 110-799, South Korea
dDepartment of Material Science and Engineering, Hallym University, Chunchon, 200-702, South Korea
eDepartment of Mechanical and Aerospace Engineering, Seoul National University, Seoul 151-744, South Korea
fBig Data Institute, Seoul National University, Seoul 151-742, South Korea
gDepartment of Bionano Engineering, Hanyang University, Ansan, 426-791, South Korea. E-mail: jwh@hanyang.ac.kr; Tel: +82-31-400-5206
hInter-university Semiconductor Research Center, Seoul National University, Seoul 151-744, Republic of Korea
First published on 27th July 2015
Abstract
A multilayer micro/nanofluidic device was presented for the selective preconcentration and online collection of charged molecules with different physicochemical properties based on ion concentration polarization phenomena. With a balance of electroosmotic drag force and electrophoretic force on the molecules, a sample mixture of sulforhodamine B and Alexa Fluor 488 could be highly preconcentrated and separated simultaneously. A repeated microchamber structure was employed to capture each dye at a desirable position. For subsequent on-chip or off-chip application, pneumatic microvalves were integrated and selectively collected the target dyes with cyclic valve operations. Using the integrated system, Alexa Fluor 488 was solely collected (with a separation resolution of 1.75) out of the mixture at a 30-fold preconcentration ratio. This integrated device would be a key component for lab on a chip applications.
1. Introduction
Recently, the micro Total Analysis System (mTAS) has been tremendously researched in the fields of analytical chemistry, diagnosis, environmental and nuclear engineering.1,2 The major components in such systems are a separator for multi-component analytes, a preconcentrator for the detection of low abundant molecules, and a sample collector for post processing. A number of studies have been performed on the preconcentration or separation of molecules using capillary electrophoresis,3 microfluidic field-amplified sample stacking,4 isoelectric focusing5 and isotachophoresis.6 In practical processes, these methods have several difficulties; (1) these methods require an additional extraction system of preconcentrated and separated molecules because they employ a free flow concept3–6 (i.e. the target compounds are migrating and concentrating simultaneously along the specific path where the background fluid is flowing), limiting subsequent processes. (2) Alternating injection of samples that should be isolated from one another causes considerable cross contamination.6 (3) While the reliability of the detection level is enhanced by increasing the concentration of samples, intricate channel geometries or specific chemicals are often required for such an experimental setup.3,5 (4) Even after successful separation and preconcentration, online detection followed by these operations can still cause an inevitable dispersion.3–6 These problems tend to increase the complexity of operations or produce false negative and false positive results, resulting in unfeasible on-chip processes. Therefore, a new design with simple structure and easy fabrication is demanded for performing fast and accurate separation and preconcentration. Furthermore, a connection to conventional off-chip analysis systems or an integration with on-chip analysis parts is expected for the ideal platform of mTAS.As a method to preconcentrate analytes, nanoscale electrokinetic phenomenon caused by the selective transportation of counter-ions in the electrolyte near a nanoporous membrane, called ion concentration polarization (ICP), has been suggested and the preconcentration factor reached up to million-fold.7 The target analytes are concentrated at pinned position, while background fluid is still flowing in ICP operation. Recently, simultaneous separation and preconcentration (or selective preconcentration) was successfully performed using ICP, demonstrating the separation of phosphorylated and unphosphorylated substrates8 or tagged and untagged DNA molecules.9 They indirectly proved that one can utilize the ICP concept not only to preconcentrate low abundant molecules but also to separate molecules depending on different physicochemical properties. However, these ICP demonstrations still have several limitations. (1) First of all, the identification of the location where reactions occur becomes labor-intensive and time-consuming tasks since the location of preconcentrated sample plug keeps uncontrollably fluctuating by the strong instability of electrokinetic flow inside ICP layer.10,11 (2) The extraction of the preconcentrated analytes for post processing without irresistible dispersion still has not been accomplished. Turning off an applied electric field let the highly preconcentrated plugs disperse quickly since there is a huge concentration gradient at the boundary. To resolve this problem, an integration with two-phase droplet generator12 or pre-binding on-site reaction13 was reported, but an additional recovery process was needed or aggressive washing steps should have been excluded, respectively. These factors significantly hinder the commercialization of further engineering developments.
To evolve ICP concept as a practical tool to selectively preconcentrate charged molecules, the integral mechanism is unprecedentedly required of (1) the stable formation of the highly preconcentrated single analyte and (2) on-demand extraction to external systems without undesirable dispersion. In this work, by designing narrow channels between repeated microchambers, the fluidic instability due to amplified electric field inside ICP was able to be suppressed. Limiting the ever-expanding ICP zone by microstructures14–17 or by external hydrodynamic flow injection18 has been reported to restrict the hydrodynamic instability and the role of vortices near the membrane.19,20 These narrow microchannels between repeated microchambers presented in this work are expected to perform in the same manner. Subsequently, pneumatic microvalve system21,22 was employed to isolate the highly concentrated sample from the original sample mixture without further dispersion, and also to collect sample plugs so that they can be further used in conventional analytical systems in either on-chip or off-chip formats.
2. Experimental section
2.1 Design of PDMS microchips
Fig. 1(A) showed the microscopic image of the multilayered selective preconcentration device with pneumatic valve system. We implemented microchamber structures repeatedly to suppress fluidic instability and differentiate the flow velocity within a single channel. Buffer channel was connected to the main channel with a perm-selective Nafion membrane. The depth of all microchannels was fixed to 15 μm and three microchambers (150 μm × 150 μm) were connected by 15 μm × 225 μm microchannel as shown in Fig. 1(A). At the right most microchamber, a microchamber with the same size was upwardly connected to collect isolated preconcentration plugs. There were fundamental and practical reasons to set the narrow height as 15 μm. In current design, the microchamber structure has the repeated height of 150 μm (wide height) and 15 μm (narrow height) in order to keep 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The numerical simulation in ESI† taught us that 10:1 was enough to focus the electric field and keep the position of the plug. Here, the wide height over 150 μm would give disperse preconcentrated plug so that we set the maximum wide height as 150 μm. The practical reason was that a mask resolution under 15 μm cost 5 times more expensive than the mask resolution over 15 μm, leading to a hurdle for commercialization. By both reasons, we kept 150 μm:15 μm in current design. A rib-shaped channel was also subsequently connected to be used as a measurement window. Microvalves (the width and height were 290 μm each) were designed to cover 4 locations as indicated with red ink in Fig. 1(A). Note that each microchamber could have its own collection channel depending on the location where target analytes were preconcentrated. However, in this work, we designed the device with only one collection channel to show the possibility of achieving the collection of selectively preconcentrated dyes. If one would like to collect dyes from different microchambers, one could install additional collection channels to the microchamber.
:1 ratio. The numerical simulation in ESI† taught us that 10:1 was enough to focus the electric field and keep the position of the plug. Here, the wide height over 150 μm would give disperse preconcentrated plug so that we set the maximum wide height as 150 μm. The practical reason was that a mask resolution under 15 μm cost 5 times more expensive than the mask resolution over 15 μm, leading to a hurdle for commercialization. By both reasons, we kept 150 μm:15 μm in current design. A rib-shaped channel was also subsequently connected to be used as a measurement window. Microvalves (the width and height were 290 μm each) were designed to cover 4 locations as indicated with red ink in Fig. 1(A). Note that each microchamber could have its own collection channel depending on the location where target analytes were preconcentrated. However, in this work, we designed the device with only one collection channel to show the possibility of achieving the collection of selectively preconcentrated dyes. If one would like to collect dyes from different microchambers, one could install additional collection channels to the microchamber.
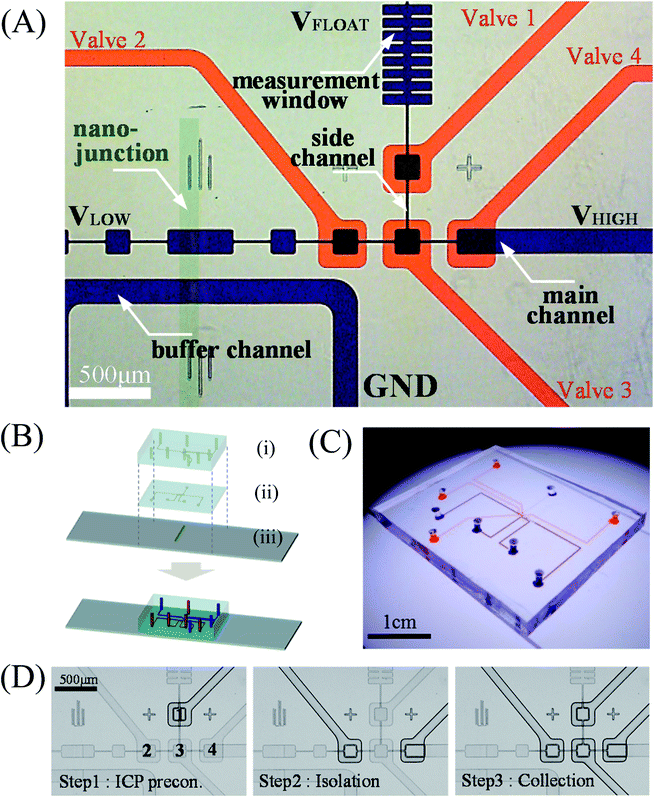 | ||
| Fig. 1 (A) Microscopic view of the selective preconcentration device. The microchannels in the ICP layer were indicated with blue and the microchannels in the valve layer were indicated red. (B) An exploded view of the device with (i) a valve layer, (ii) an ICP layer and (iii) a Nafion patterned slide glass. (C) Assembled multilayered online selective preconcentration device. (D) Microscopic images of 3-step valve control sequence: Step 1 – ICP preconcentration to form a highly concentration plug, Step 2 – isolation of a highly preconcentrated plug at specific microchamber, and Step 3 – collection of the highly preconcentrated plug into upward channel. See ESI Video.† | ||
2.2 Materials and reagents
For a main channel solution, 5 mM KCl solution (Sigma Aldrich, USA) was used with addition of two fluorescent dyes, sulforhodamine B (SRB) (24.14 nM, Sigma Aldrich, USA) and Alexa Fluor 488 (Alexa) (0.90 μM, Invitrogen, USA). Their separation and preconcentration dynamics were able to be measured and identified with two different fluorescent colors of orange (SRB) and green (Alexa). For a buffer channel solution, 100 mM KCl was used to minimize a voltage drop in the buffer channel. The ICP operation has been well performed in high salinity samples over 100 mM.23,24 In this work, 5 mM KCl was chosen under assumption that the real sample could be post-processed such as removing large molecules and cells (dilution). In such case, the solution of 5 mM concentration would be a proper standard model.2.3 Preparation of poly(dimethylsiloxane) (PDMS) microchips
A 4-inch wafer was spun to coat 15 μm layer of SU-8 2015 photoresist (MicroChem. Inc, USA) at 500 rpm of 10 seconds (pre-spinning) and at 3250 rpm for 30 seconds (spinning). After 3 minutes of soft bake at 95 °C, the layer was exposed two times to ultraviolet (UV) of 365 nm wavelength for 23.3 seconds (400 mW) using a mask aligner. Post bake was for 4 minutes at 95 °C, then was soaked in isopropyl alcohol (IPA) for 3 minutes. For easy detachment of the PDMS device, trichloro(1,1,2,2-perfluorooctyl)silane was applied to the master wafer with a vacuum assisted deposition desiccator.PDMS base and curing agent (Sylgard 184 Silicone elastomer kit, Dow Corning, USA) were mixed at 10:1 ratio and desiccated in a vacuum pump for 1 hour to remove air bubbles. The mixed solution was poured onto the master and cured in room temperature overnight for the valve layer (Fig. 1(B)-(i)), preventing alignment error due to thermal shrinkage. For the ICP layer, the mixed solution was poured onto the patterned 4-inch wafer and spun at 1500 rpm for 300 seconds (JSP4A, JD Tech, Korea) to coat 25 μm PDMS layer (Fig. 1(B)-(ii)). Then, the coated wafer was placed on a hot plate for 30 minutes at 95 °C. The PDMS replicas were peeled off from the master and the inlet and outlet holes were punched out.
A polymeric nanoporous material, Nafion (20 wt% resin, Sigma Aldrich, USA), was used for the nanojunction between the microchannels. A single strip of Nafion (100 μm wide and 1 cm long) was patterned on a slide glass using a PDMS piece that has a single straight microchannel and was heated at 95 °C for evaporating solvent after removing the PDMS piece (Fig. 1(B)-(iii)).18 Then, each layer was bonded to the Nafion patterned slide glass by oxygen plasma bonder (Femto Science, Korea) under an alignment through the stereo microscope (SZ61, Olympus, Japan) creating a multilayer device. Final assembled micro/nanofluidic device is shown in Fig. 1(C).
2.4 ICP operation
Chemical samples were loaded into the main and buffer microchannel by applying positive pressure from each inlet reservoir. Ag/AgCl electrodes were inserted into the inlet and buffer reservoirs on the device and connected to a power supply (Keithley 6517 and Keithley 238, Keithley Instruments, USA). For applying the electric field, an independent voltage control was required on each of the four reservoirs. For the continuation on separation and preconcentration, 60 V and 90 V were applied to the left and the right reservoir of the main channel, respectively, while the reservoir of the buffer channel was grounded. The motions of fluorescent dyes were tracked by an inverted fluorescence microscope (IX53, Olympus, Japan) and recorded by CellSens (Olympus, Japan) computer program.2.5 Valve operation
A compressor (Gfrog, Stylex, Korea) as a pressure source was connected to 4 mini solenoid valves (SY 3120, SMC pneumatic, Korea) and a polystyrene tube connected a solenoid valve and an inlet of valve layer in the PDMS device. Before the connection, DI water was filled into the microchannels of valve layer to reject air bubbles into the ICP layer. In order to manipulate the pneumatic microvalves automatically, a customized LabVIEW code (National Instrument, USA) and a DAQ board (USB-DAQ 6341, National Instrument, USA) were used.The valve operation consisted 3 steps as shown in Fig. 1(D). Step 1 (ICP preconcentration): valve 1 was closed when selective preconcentration was in process. Step 2 (isolation): when the targeted preconcentration plug reached the microchamber which was connected to the side channel, valve 1 was opened and valve 2 and 4 were closed to isolate the sample plug from the main channel. Step 3 (collection): then, by closing valve 1 and 3 while valve 2 and 4 were still closed, the isolated plug was squeezed into the measurement window. Fluorescent dyes diffused into a larger area and the fluorescence intensity was analyzed without being saturated. By leaving valve 1 closed and others opened, the whole 3-step process cyclically repeated for further collections. See ESI Video.† One can control the duration of Step 1 to obtain a different preconcentration ratio.
Three microchambers with valves were basic building blocks under our current system. If more than 2 analytes are needed to be preconcentrated and separated, the number of microchambers and valves could be added. To do this, the number of microchamber should be larger than the number of analyte. For a different kind of dyes rather than SRB and Alexa, one can tune the electric field to fill targeted microchamber at one's discretion. Here the three-microchamber system was enough for selective preconcentration of 2 dyes regardless of the types of dye.
3. Results and discussions
3.1 Demonstration of selective preconcentration
Fig. 2(A) demonstrated that two different dyes were simultaneously preconcentrated and separated over time. See ESI Video.† Without electric field along the nanojunction, no fluorescent dyes were observed in the channel because the initial concentrations of dyes were too low to be detected. As we applied 90 V for VHIGH and 60 V for VLOW on each end of the main channel with grounded buffer channel, ICP was triggered near the membrane. Micro air bubbles could be generated at the electrode surface with O(10) nA operation currents. Even with these relatively high applied voltages, most of electrical potential was dropped inside the ion depletion zone. In addition, because the electrodes were inserted at the reservoirs which located far from the nanojunction and the reservoirs were opened to ambient atmospheric pressure, the bubble at the electrode surface can float and disappear into the air so that the selective preconcentration operation was not interfered by the bubbles. As reported in previous researches,25 the ion depletion zone caused significant and dynamic perturbation in a local ion concentration and in an amplified electric field near the membrane along with the generation of strong electrokinetic flow. The high electric field gradient at the boundary of depletion zone rejected the penetration of charged molecules into the ion depletion zone so that both dyes stacked and detected at the boundary.7 | ||
| Fig. 2 (A) Time-lapse images of selective preconcentration of SRB and Alexa. High concentration ratio was represented by the brightness of fluorescence. Since the physicochemical properties of two dyes were different, they were preconcentrated at different locations. See ESI Video.† (B) Schematic diagram of selective preconcentration mechanism with different mobility. The separation was drawn by the equilibrium between electroosmotic drag force and electrophoretic force exerted on molecules. | ||
At the beginning of selective preconcentration, both dyes were preconcentrated in the microchamber 1. After the ion depletion zone became stabilized over time, the preconcentrated plugs of two dyes with different mobility were separated with a separation resolution26 of 1.75. We have adopted the definition of the separation resolution as Rs = (peak to peak distance)/(average width of bands) leading to Rs of 1.75 (525 μm/((450 μm + 150 μm)/2)). This number indicates a perfect separation (i.e. no overlap) in case of Gaussian distribution. While there has been no exhaustive theoretical analysis on this selective preconcentration process, the mechanism of selective preconcentration was empirically analyzed using force balance.8,27 The force is mobility times particle velocity28,29 so that we would like to discuss the balance in terms of force rather than mobility, since the size of molecules (∼1 nm) were similar. This separation was drawn by the equilibrium between an electroosmotic drag force and an electrophoretic force as described in Fig. 2(B).8 The electroosmotic drag exerted on the molecules was Fdrag = 6πμRu where μ is the dynamic viscosity, R is the radius of the spherical object, and u is the electroosmotic velocity of bulk which is independent from molecule's properties, while the electrophoretic force exerted on the charged species was FEP = qE where q is a net electric charge and E is an electric field. According to an experimental observation shown in Fig. 2(A), Alexa (green) moved toward the right reservoir, while SRB (orange) stayed near the depletion zone boundary. Since Fdrag is mainly determined by R which is almost the same for SRB and Alexa, the value of net charge played a key role in determining the equilibrium positions balanced by Fdrag and FEP. This argument was in line with the experimental result since the net charge of SRB and Alexa is −1 and −2, respectively.
3.2 Stabilization of selective preconcentration
While the phenomenon of different equilibrium positions of molecules with different mobility has been already reported in previous researches,8,27 one strong plug and one smaller plug in parabolic shape were reported and the locations of preconcentrated plugs were always unpredictable due to the strong instability by local amplified electric field near the nanojunction,25 instead of stably generated plugs with high concentration. Therefore, it was important to control the positions of the concentrated sample plugs and to stabilize the ion depletion zone. In our system, the repeated narrow and wide channels was able to stabilize the depletion zone by restricting the expansion of vortices and strong electrokinetic flows in the ion depletion zone,15,16,19,20,30,31 thereby forming a plug at desirable positions. Details about theoretical and experimental analysis can be found in ESI.† Briefly, the repeated microchamber structures were able to effectively confine the ion depletion zone in comparison to the straight channel. This meant that the ICP layer in the repeated chamber would be stabilized because the flow instability generated by concentration fluctuation32 occurred only inside the first chamber where nanojunction was installed. While monotonically increased electric potentials were observed in straight microchannel, there was a (almost) plateau inside microchamber with steep changes in narrow regions. A charged species would be immobilized in this plateau region. Due to the pseudo-periodic electric field and the plateau of electric potential, the preconcentrated plug would be easily trapped in each chamber. Consequently the control of each accumulated molecules' location became simple and tailing effect was reduced. This alternating geometry also differentiated the velocity of the fluidic flow at the boundary between the narrow and the wide channel, and therefore, it increased the efficiency of separation and helped the membrane microvalve system for further blocking and squeezing of the analytes.3.3 Quantitative analysis for separation and preconcentration efficiency
The selective preconcentration was quantitatively analyzed by measuring pixel intensity with fixed exposure time. The intensities of the original sample and reference solutions at different concentrations were compared to accurately measure the preconcentration ratio, since the pixel intensity was not linearly proportional to the concentration of dyes. As shown in Fig. 3(A), the average pixel intensities of dyes increased in each microchamber in order. The both dyes (Alexa + SRB) were rapidly preconcentrated in the microchamber 1 from the initial to 10 minutes. From 10 minutes to 25 minutes, the green plug (Alexa) moved to the microchamber 2 while the intensities of the plugs simultaneously increased. This agreed with the microscopic image in Fig. 2(A) that the orange plug (SRB) stayed in the microchamber 1, while the green plug (Alexa) moved to the microchamber 2. After 25 minutes, the green plug (Alexa) moved to the microchamber 3, while the average pixel intensity of the microchamber 2 sharply diminished. In this point, the average pixel intensity of the microchamber 2 did not decrease to zero. This was because the orange plug (SRB) was saturated in the first microchamber so that it went on to the microchamber 2. In the microchamber 3, the average pixel intensity reached up to 98.2, which denoted that the green plug (Alexa) was preconcentrated above 100-fold since the average pixel intensities of 10×, 50×, and 100× concentrations of Alexa Fluor 488 were 12.9, 58.9, and 95.3 respectively. After 30 minutes, the repeated microvalve operations were started, showing severe fluctuations and this part will be discussed later.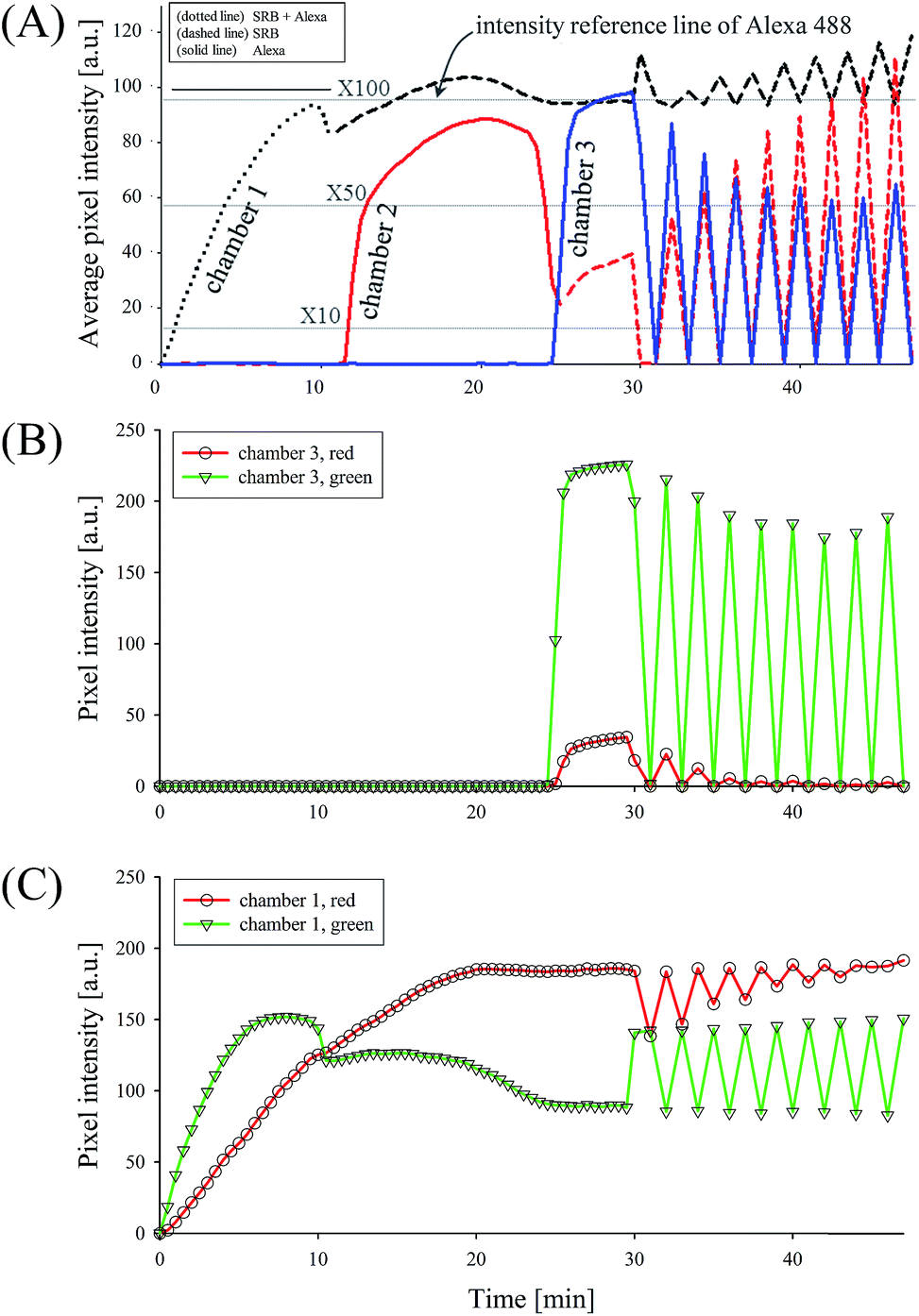 | ||
| Fig. 3 (A) Overall fluorescence intensity tracked in each microchamber as a function of time. Dotted line, dashed line and solid line represented the mixture of SRB with Alexa, only SRB and only Alexa, respectively. Three different colors indicated the pixel intensity of each microchamber (black for microchamber 1, red for microchamber 2 and blue for microchamber 3). The concentration ratio of selectively preconcentrated Alexa in microchamber 3 exceeded 100-fold, maintaining the preconcentration factor high enough in repeated valve operations. (B) Red/green color profile in microchamber 3, showing the microchamber 3 only had Alexa. (C) Red/green color profile in microchamber 1, showing the microchamber 1 only had SRB. Fluctuations after 30 minutes in all plots showed that the preconcentration factor maintains over 50-fold along the cyclic valve operations. While the error bars were omitted for clear visibility, the tests were conducted at least 10 times with different devices for the repeatability and reliability. | ||
In previous researches, the two separated plugs have been distinguished only by their positions since only one dye was tagged for two or more different kinds of molecules.8,27 Compared to these works, RGB color profile was rigorously investigated to validate the separation of two molecules. Since the emission from SRB has both red and green color, it was efficient to measure color intensities from microchamber 3 as the indicator of separation, where only Alexa dye was preconcentrated. Since the pixel intensities of blue and red were almost the same, only red and green colors were discussed. The color profile of 100-fold reference solution of Alexa at 0.90 μM was 33.28 in red and 219.76 in green under the exposure time of 300 ms. As shown in Fig. 3(B), the pixel intensity of the microchamber 3 in the main channel was 223.20 in green and 31.17 in red at the operation time from 25 minutes to 30 minutes, which agreed with the reference values of the 100-fold, meaning that the majority occupying the microchamber 3 was only Alexa dye. In addition to this, the color profile of the microchamber 1 (Fig. 3(C)) contained high value of red color which was not found in the microchamber 3 as shown in Fig. 3(B). The ratio of red and green color was reversed at 10 minutes and maintained around 2:1 from 25 minutes to 30 minutes in the microchamber 1. Since SRB was dominant in the original solution (SRB at 24.14 nM + Alexa at 0.90 μM) and its red/green ratio was 2:1, we concluded that the microchamber 1 was solely occupied by SRB after 25 minutes. This color profile measurement provided an accurate platform to measure the separation efficiency between two dyes with different mobility.
3.4 Collection of preconcentrated plugs by valve system
As the operation continued further, the preconcentration factor increased accordingly. However, subsequent process using selectively preconcentrated analytes was difficult since the selective preconcentration was performed while the background solution freely flows in a single channel. Besides, a subsequent on-chip process which should exclude electrical operation was inapplicable because the plug drastically dispersed as shown in Fig. 4(A) when the applied voltage was turned off. To resolve this problem, a plug was required to be isolated and transported to another site, while the preconcentration ratio maintained without loss for either on-chip or off-chip applications. This was able to be realized using pneumatic microvalve system. To guarantee the role of valves to block the plug from the original sample channel, the extent of dispersion was compared between the case of valve OFF and valve ON as described in Fig. 4(A) and (B). See ESI Videos.† | ||
| Fig. 4 Time-lapse images showing the dispersion of isolated plug when the valves were (A) OFF and (B) ON. See ESI Videos.† | ||
The valves were operated in a repeated cycle as described in Fig. 1(D) to collect the selectively preconcentrated plug into the side channel. In a cycle, the plug was isolated from the original sample solution and squeezed up into the side channel. To analyze the preconcentration factor achieved from the process, a rib-shaped measurement window was installed in the middle of the side channel. This measurement window was designed for metering how high the preconcentration ratio would be achieved through the process. In the meantime, the measurement window had larger area than the side channel itself so that it had low fluidic resistance and the side channel connected to an open reservoir to atmospheric pressure, allowing subsequent processes without any disturbance. As shown in Fig. 5(A), no fluorescence intensity was observed in the measurement window at t = 0, because the measurement window was initially filled with sample solution. By the first valve operation, the pixel intensity of measurement window already exceeded 10-fold of the initial concentration of Alexa as plotted in Fig. 5(B). With repeated valve operations, the concentration of collected plugs in the measurement window continuously increased so that the concentration reached over 30 times of the initial concentration after the sixth operation. After the ninth collection, it was observed that the measurement window was fully filled with green dyes. After this point, a diffusive mixing would not occur anymore. The error bars decreased and the signal saturated according to the number of valve operation, since we measured area averaged fluorescent intensity in whole measurement window. It led largest error bar at the 1st collection because the window was not fully filled with the dye. See ESI Video.†
 | ||
| Fig. 5 (A) Snapshots of repeated valve operations collecting selectively preconcentrated plugs. Compared to the initial state, the measurement window was being filled with green dye (Alexa) over several valve operations. See ESI Video.† (B) Average intensity change in the measurement window over number of repetition of valve operations. The average intensity exceeded 10-fold in a single valve operation, and 30-fold after nine operations. Note that there was no SRB in the measurement window. | ||
The preconcentration factors of each Step 1 in repeated cycles were maintained high enough to continuously increase the pixel intensity of the measurement window. Fig. 3(A) also supported this result. In the figure, the valves were repeatedly operated in the same cycle with the interval of 2 minutes after 30 minutes of selectively preconcentrating operation. When the analytes in the microchamber 3 were isolated by valves and moved up into the side channel, the pixel intensity of the microchamber 3 was zero. The intensity of microchamber 3 increased again with the next preconcentration operation for 2 minutes. The concentration of Alexa in the microchamber 3 was maintained over 50-fold for all valve operations. The upheld high preconcentration ratio of collected sample was led by stable and reproducible valve operations. Since microvalve systems squeezed the preconcentrated plugs by mechanical force, the mechanism of the collection process barely agitated the external electric field.
4. Conclusions
In this paper, we demonstrated a multilayer micro/nanofluidic device for selective preconcentration. The operating principle of the device was based on the high electric field gradient induced by ICP phenomenon. In the main channel, two opposing electrophoretic force and electroosmotic drag force exerted on the charged molecules were balanced depending on the physicochemical properties of the molecules. Importantly, the repeated microchamber structure suppressed the undesirable instability of the ICP operation, pinning the location where the preconcentrated plug was formed. With this device, SRB and Alexa were simultaneously separated and preconcentrated at the separation resolution of 1.75. By introducing pneumatic microvalves, dispersion due to the high concentration gradient was prevented, and Alexa plugs preconcentrated at 100-fold was able to be isolated from the main channel without the loss of the ratio during the cyclic valve operations. After all, 30-fold of Alexa was able to be collected in the measurement window. RGB signals from the preconcentrated analytes were rigorously analyzed for quantitative and qualitative separation efficiency, while previous researches relied only on the position tracking of the molecules. Given the importance of extracting selective preconcentrated analytes, we expect that this mechanism and structure of the micro/nanofluidic device would be a powerful tool for lab on a chip applications.Acknowledgements
This work is supported by Basic Science Research Program (2013R1A1A1008125), the Center for Integrated Smart Sensor funded as Global Frontier Project (CISS-2011-0031870) and Future based Technology Development Program (2012-0009563) by the Ministry of Science, ICT & Future Planning and Korean Health Technology RND project, Ministry of Health and Welfare Republic of Korea (HI13C1468, HI14C0559). Also this work partially supported by BK21 plus program.References
- D. R. Reyes, D. Iossifidis, P.-A. Auroux and A. Manz, Anal. Chem., 2002, 74, 2623–2636 CrossRef CAS.
- P.-A. Auroux, D. Iossifidis, D. R. Reyes and A. Manz, Anal. Chem., 2002, 74, 2637–2652 CrossRef CAS.
- V. Dolnik, S. Liu and S. Jovanovich, Electrophoresis, 2000, 21, 41–54 CrossRef CAS.
- B. Jung, R. Bharadwaj and J. G. Santiago, Electrophoresis, 2003, 24, 3476–3483 CrossRef CAS PubMed.
- V. Neuhoff, N. Arold, D. Taube and W. Ehrhardt, Electrophoresis, 1988, 9, 255–262 CrossRef CAS PubMed.
- B. Jung, R. Bharadwaj and J. G. Santiago, Anal. Chem., 2006, 78, 2319–2327 CrossRef CAS PubMed.
- S. J. Kim, Y. A. Song and J. Han, Chem. Soc. Rev., 2010, 39, 912–922 RSC.
- L. F. Cheow, A. Sarkar, S. Kolitz, D. Lauffenburger and J. Han, Anal. Chem., 2014, 86, 7455–7462 CrossRef CAS PubMed.
- H. Song, Y. Wang, C. Garson and K. Pant, Microfluid. Nanofluid., 2014, 17, 693–699 CrossRef CAS PubMed.
- P. Kim, S. J. Kim, K.-Y. Suh and J. Han, Nano Lett., 2010, 10, 16–23 CrossRef CAS PubMed.
- S. J. Kim, S. H. Ko, R. Kwak, J. D. Posner, K. H. Kang and J. Han, Nanoscale, 2012, 4, 7406–7410 RSC.
- C.-H. Chen, A. Sakar, Y.-A. Song, M. A. Miller, S. J. Kim, L. G. Griffith, D. A. Lauffenburger and J. Han, J. Am. Chem. Soc., 2011, 133, 10368–10371 CrossRef CAS PubMed.
- Y.-C. Wang and J. Han, Lab Chip, 2008, 8, 392–394 RSC.
- S. H. Ko, S. J. Kim, L. F. Cheow, L. D. Li, K. H. Kang and J. Han, Lab Chip, 2011, 11, 1351–1358 RSC.
- Y. Green, S. Shloush and G. Yossifon, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 89, 043015 CrossRef.
- Y. Green and G. Yossifon, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 89, 013024 CrossRef.
- S. J. Kim, Y.-C. Wang, J. H. Lee, H. Jang and J. Han, Phys. Rev. Lett., 2007, 99, 044501 CrossRef.
- I. Cho, G. Y. Sung and S. J. Kim, Nanoscale, 2014, 6, 4620–4626 RSC.
- M. Jia and T. Kim, Anal. Chem., 2014, 86, 7360–7367 CrossRef CAS PubMed.
- M. Jia and T. Kim, Anal. Chem., 2014, 86, 10365–10372 CrossRef CAS PubMed.
- J. W. Hong, V. Studer, G. Hang, W. F. Anderson and S. R. Quake, Nat. Biotechnol., 2004, 22, 435–439 CrossRef CAS PubMed.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- J. H. Lee, B. D. Cosgrove, D. A. Lauffenburger and J. Han, J. Am. Chem. Soc., 2009, 131, 10340–10341 CrossRef CAS PubMed.
- S. J. Kim, S. H. Ko, K. H. Kang and J. Han, Nat. Nanotechnol., 2010, 5, 297–301 CrossRef CAS PubMed.
- S. J. Kim, L. D. Li and J. Han, Langmuir, 2009, 25, 7759–7765 CrossRef CAS PubMed.
- J. C. Giddings, Unified Separation Science, John Wiley & Sons, Inc, New York, NY, 1991 Search PubMed.
- L. F. Cheow and J. Y. Han, Anal. Chem., 2011, 83, 7086–7093 CrossRef CAS PubMed.
- J. Happel and H. Brenner, Low Reynolds Number Hydrodynamics: With Special Applications to Particulate Media, Springer, Netherlands, 1983 Search PubMed.
- S. Kim and S. J. Karrila, Microhydrodynamics: Principles and Selected Applications, Dover Publications, 2013 Search PubMed.
- S.-H. Lee, H. Lee, T. Jin, S. Park, B. J. Yoon, G. Y. Sung, K.-B. Kim and S. J. Kim, Nanoscale, 2015, 7, 936–946 RSC.
- S. Nam, I. Cho, J. Heo, G. Lim, M. Z. Bazant, D. J. Moon, G. Y. Sung and S. J. Kim, Phys. Rev. Lett., 2015, 114, 114501 CrossRef.
- C. L. Druzgalski, M. B. Andersen and A. Mani, Phys. Fluids, 2013, 25, 110804 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12639h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |