
Thermally stable phthalonitrile resins based on multiple oligo (aryl ether)s with phenyl-s-triazine moieties in backbones†
Abstract
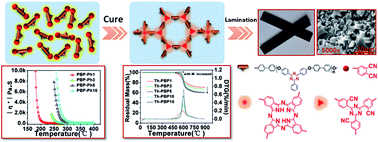
Many efforts have been devoted to tailoring the architecture of phthalonitrile (PN) polymers in recent years. Herein, we disclose a series of novel PN oligomers (PBP-Phs) bearing heteroaromatic phenyl-s-triazine moieties, serving as thermally stable segments in the polymer backbones. With bis(4-[4-aminophenoxy]phenyl)sulfone as a curing additive, PBP-Phs displayed commendable processability. After curing at high temperatures (up to 375 °C), the resulting networks (Th-PBPs) exhibited high glass transition temperatures ranging from 294 °C to 400 °C and outstanding thermal stability with a weight retention of 95% in N2 lying between 538 °C and 582 °C; the overall thermal properties were closely connected with the oligomer molar weights. Additionally, the feasibility of producing Th-PBPs reinforced with unidirectional continuous carbon fibers (CF) was evaluated. The results show that CF/Th-PBP laminates possess high flexural strength (1339–1855 MPa) and interlaminar shear strength (71.8–92.2 MPa).
 Please wait while we load your content...
Please wait while we load your content...

