
Smooth CH3NH3PbI3 from controlled solid–gas reaction for photovoltaic applications†
Abstract
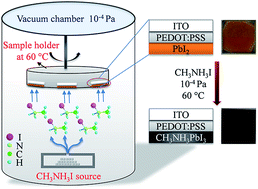
The merits of high power conversion efficiency (PCE) and easy preparation make organic–inorganic perovskite solar cells one of the most promising solar devices. However, PCE is greatly dependent on the morphology of perovskite thin film. Here, we report a solid–gas reaction method to fabricate very smooth CH3NH3PbI3 thin film with high coverage. Through controlling the reaction rate between CH3NH3I and PbI2 by tuning the PbI2 substrate temperature and the evaporation rate of CH3NH3I, we obtain a CH3NH3PbI3 layer with roughness of 7.37 nm. Besides, no post-treatment annealing is needed after film formation using our approach. With about 250 nm perovskite active layer, the solar cells exhibit a PCE of 10.0% with little hysteresis.
 Please wait while we load your content...
Please wait while we load your content...

