
Graphitic-carbon nitride support for the synthesis of shape-dependent ZnO and their application in visible light photocatalysts†
Pragati Fageriaa,
Roshan Nazira,
Subhashis Gangopadhyayb,
Harish C. Barshiliac and
Surojit Pande*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Rajasthan 333031, India. E-mail: spande@pilani.bits-pilani.ac.in; surojitpande@gmail.com; Fax: +91-1596-244183; Tel: +91-1596-515709
bDepartment of Physics, Birla Institute of Technology and Science, Pilani, Rajasthan 333031, India
cSurface Engineering Division, CSIR-National Aerospace Laboratories, Bangalore 560017, India
First published on 15th September 2015
Abstract
Shape-dependent synthesis of ZnO has been developed on the surface of g-C3N4 following a simple and reproducible strategy. Initially, graphitic-carbon nitride (g-C3N4) was synthesized by pyrolysis of urea which was further used to grow ZnO nanostructures via refluxing conditions. Different hydrolyzing agents, such as hexamethylenetetramine (HMT) and ammonia were used to synthesize dumbbell and cone structures, respectively. Apart from hydrolyzing agents, cetyltrimethylammoniumbromide (CTAB) was also used as a growth controlling agent. Structural, morphological and optical characterizations of the as-synthesized materials were performed by using FESEM, TEM, XRD, XPS, UV-vis etc. techniques. After successful synthesis, the as-synthesized heterostructures were explored as visible light driven photocatalysts towards organic pollutant (methylene blue and phenol) degradation. The photocatalytic performances of bare ZnO, dumbbell and cone structures of g-C3N4/ZnO as well as g-C3N4, have been examined thoroughly. Photocatalytic results revealed that g-C3N4/ZnO heterostructures exhibit a higher efficiency under the illumination of visible light as compared to pure g-C3N4. Superior photodegradation activity of the g-C3N4/ZnO heterostructure originated from the synergistic effect and high charge separation at the interface of g-C3N4 and ZnO has also been discussed.
Introduction
Carbon nitride has attracted enormous attention in the field of material science, since Liu and Cohen guessed that carbon and nitride are good applicants for the intense rigidity of materials.1 The unique layered structure and a characteristic electronic band structure of carbon nitride (C3N4) make it stable at ambient conditions. Owing to its exclusive physicochemical properties, such as rigidity, light weight, chemical inertness, water resistivity, and biocompatibility, C3N4 has been regarded as a promising material for surface functionalization, light emitting devices, and metal free electrocatalysis and photocatalysis.2–7 Among the various allotropes of polymeric C3N4, graphite like carbon nitride (g-C3N4) is a promising and most stable allotrope. Graphitic (g)-C3N4 as an analogue of graphite has attracted considerable interest because of its suitable band alignment for water splitting, selective oxidation reactions, and environmental pollutant degradation.2,8–10 Interestingly, the matrix of g-C3N4 can offer active sites for binding with many organic/inorganic compounds or metals, which supply a convenient route to functionalize g-C3N4 surface with high reactivity.8,11–13g-C3N4 represents an important class of conjugated polymeric semiconductors.14 Similar to graphene, g-C3N4 possesses π-conjugated electronic structure with a band gap of 2.7 eV.2–5,8,9,15 g-C3N4 is a layered metal-free semiconductor with an interlayer distance of about 0.33 nm (single layer of g-C3N4).6 Bulk g-C3N4 can easily be prepared by polycondensation reaction of organic precursors, e.g., urea, cyanamide, dicyandiamide, and melamine. Liu and co-workers13 demonstrated a large-scale production of g-C3N4 by pyrolysis of urea using different reaction temperatures and times. They have also reported the photocatalytic activity of g-C3N4 in dye degradation using visible light.13 Another simpler approach for the synthesis of polymeric g-C3N4 using urea as precursors and their application in rhodamine B (RhB) dye degradation using visible light has been reported by Dong and co-workers.12 Surfactant (Pluronic P123) assisted synthesis of porous g-C3N4 using melamine has been reported by Yan and co-workers, where they found a significant improvement in the photocatalytic H2 evolution from water using visible light.16 Wang and co-workers9 reported chemically and thermally stable polymeric carbon nitride, a metal free photocatalyst with high capability, to generate hydrogen from water. Additionally, they have modified the carbon nitride with small amount of Pt metal to vary the hydrogen production with better activity and reduced experimental error. However, a low quantum efficiency for Pt modified C3N4 surface has also been observed.9
The photocatalytic performance of bare g-C3N4 is limited due to the high recombination rate of the photogenerated electron–hole pairs.17–21 This restricted photocatalytic performance of g-C3N4 can be enhanced by synthesizing porous structures,20–23 doping and/or coupling of g-C3N4 with transition metals, metal oxides and sulphides, and active protonation.19,24–28 To increase the separation between photogenerated electron–hole pairs and to promote the photocatalytic activity, researchers have coupled g-C3N4 with ZnO, TiO2, CdS, ZnWO4, BiPO4, Bi2WO6, and Ag2O semiconductors.20–29 Hence, efforts have been given to suppress the rate of recombination of charge carriers and improving the consumption of solar light, by combining two semiconductors. ZnO is an UV-light active and large exciton binding energy (60 MeV) material with Eg = 3.37 eV that also limits the practical application in visible light photocatalysis.30 Sun and co-workers20 reported a calcination process for the synthesis of a composite g-C3N4–ZnO as visible light photocatalysts with variable amount of ZnO, which they have utilized for methyl orange and p-nitrophenol photodegradation using visible light with 3–6 times better activity as compared to bare g-C3N4.20 The improved photocatalytic activity and optimum synergistic effect of ZnO hybridized g-C3N4 has also been reported by Wang and co-workers.21 Synthesis of core–shell nanoplates of N-doped ZnO/g-C3N4 via ultrasonic dispersion process and their excellent visible light photocatalytic activity has been reported by Kumar and co-workers,30 which was due to the less electron–hole pair recombination at the interface of N-doped ZnO/g-C3N4. Liu and co-workers31 reported the synthesis of g-C3N4/ZnO composite by deposition–precipitation technique, which has been used as a visible light active photocatalyst with a first-order kinetics for both photooxidation and photoreduction reaction. Synthesis of g-C3N4/ZnO nanotube by electrochemical methods and their improved photoelectrochemical properties due to synergistic effect between g-C3N4 and ZnO has been reported by Wang and co-workers.32 All these reports demonstrate a heterostructure semiconductors, combination of ZnO and g-C3N4, having eminent photocatalytic performance under the illumination of visible light. Being inspired from the above studies we have synthesized morphology-dependent ZnO structures on g-C3N4 surface. Best of our knowledge, here we are reporting for the first time the shape-dependent synthesis of ZnO on g-C3N4 surface and their application in photocatalysis.
Within this work, we report a simple and facile approach to synthesize visible light active heterostructures of g-C3N4 and ZnO, via chemisorption of zinc precursors followed by hydrolysis and dehydration. Firstly, bulk g-C3N4 was synthesized followed by the formation of dumbbell and cone shaped ZnO on its surface using CTAB and different hydrolyzing medium. The analytical techniques like UV-vis, powder X-ray diffraction (PXRD), Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), transmission electron microscopy (TEM), energy dispersive spectroscopy (EDS), and X-ray photoelectron spectroscopy (XPS) were used as characterizing tools to corroborate the synthesis of bulk g-C3N4 and g-C3N4/ZnO composite. Methylene blue (MB) dye and phenol were chosen as model compounds to evaluate the photo-activity of the as-synthesized catalysts under the illumination of visible light. g-C3N4/ZnO heterostructures show superior activity as compared to bare g-C3N4 as well as pure ZnO nanoparticles. Due to type-II band alignment of ZnO and g-C3N4, interface between g-C3N4 and ZnO helps for faster transportation of photogenerated charge, which results g-C3N4/ZnO to be an efficient photocatalyst. Finally, the reusability of g-C3N4/ZnO has also been executed to prove the stability of catalyst.
Experimental procedure
Chemicals
All chemicals were used as purchased without any further purification and of analytical grade (AR). Chemicals from different suppliers such as, Urea extrapure (Sd-fine chemicals), zinc sulphate (ZnSO4·7H2O, SD fine-chem limited), cetyltrimethylammonium bromide (CTAB, Spectrochem Pvt. Ltd.), hexamethylenetetramine (HMT, Alfa aesar), ammonia solution with sp. gr. 0.91 (SD fine-chem limited), methylene blue (Sigma Aldrich), and phenol (SD fine-chem limited) were used. Millipore water was used throughout the experiment.Synthesis of bulk g-C3N4
Synthesis of bulk g-C3N4 was reported elsewhere.13–15 Briefly, bulk g-C3N4 was synthesized by thermal treatment of 10.0 g urea in a covered silica crucible under ambient air pressure. Initially, urea powder was dried at 80 °C for an hour on water bath and then the crucible with urea was placed in the muffle furnace at 550 °C for 2.5 hour keeping the heating rate of 10° min−1. Finally, the yellow colour product was collected at room temperature and washed with nitric acid (0.1 M) four times to remove the extra impurities (e.g., NH3 etc.), which was formed during the reaction and dried at 80 °C.Synthesis of dumbbell and cone structure of ZnO on g-C3N4 surface
The as-prepared bulk g-C3N4 powder was used as a support for the synthesis of ZnO dumbbell. Initially, 50 mg of bulk g-C3N4 powder was sonicated for 30 min in 50 mL of water for complete dispersion. Now, to the aqueous dispersion of g-C3N4, 2.5 mL of 10−1 M ZnSO4·7H2O and 2.5 mL of 10−1 M CTAB were added in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with manual stirring. The pH of the above solution was set up to alkaline (pH ∼ 7.5) using hexamethylenetetramine (HMT) (5 mL of 10−1 M was added) solution with continuous stirring. Subsequently, the reaction mixture was kept at 90–95 °C for 6 h in reflux condition to complete the formation of ZnO dumbbell on the surface of g-C3N4. The white powder was collected and washed five times using methanol and dried in air.
:1 ratio with manual stirring. The pH of the above solution was set up to alkaline (pH ∼ 7.5) using hexamethylenetetramine (HMT) (5 mL of 10−1 M was added) solution with continuous stirring. Subsequently, the reaction mixture was kept at 90–95 °C for 6 h in reflux condition to complete the formation of ZnO dumbbell on the surface of g-C3N4. The white powder was collected and washed five times using methanol and dried in air.
g-C3N4/ZnO cone heterostructure was synthesized using the same procedure described for g-C3N4/ZnO dumbbell, except ammonia solution was used to maintain the alkaline (pH ∼9–10) instead of HMT solution.
Photocatalysis
For photocatalytic activity of the as-synthesized materials, 1 mL of 3 × 10−4 M MB solution and 5.0 mg of g-C3N4/ZnO catalysts (dumbbell and cone) were taken in 9 mL water so that the final concentration of MB was 3 × 10−5 M in 10 mL water. Prior to irradiation, the aqueous solution was stirred continuously for ∼20 min in the dark to confirm the saturation of dye on g-C3N4/ZnO catalyst surface. After saturation, the reaction mixture was placed under visible light with constant stirring to initiate the degradation reaction. The source of visible light was a tungsten bulb of 60 W, which emits mainly visible light. At regular irradiation time or interval (∼20 min), 3.0 mL of aliquot was taken from the reaction mixture, centrifuged at 1000 rpm for UV-vis measurement, and re-added carefully (3.0 mL solution and catalyst) to the original reaction container. Photodegradation kinetics of MB dye was monitored by observing the change in absorbance intensity at 664 nm (maximum intense peak for MB dye) with time by UV-vis spectroscopy.Similar procedure was applied with phenol degradation using visible light, 0.4 mL of 5 × 10−3 M aqueous phenol solution and 5.0 mg of catalyst were used for the decomposition reaction so that the final concentration of phenol in 10 mL water was 2 × 10−4 M.
For MB and phenol dye % degradation or degradation yield was calculated using initial and time-dependent absorbance values:

Optical characterization
Ultraviolet-visible diffuse reflection spectra were recorded on a Jasco V-650 Spectrophotometer (model no. UV-1800) with a deuterium and tungsten-halogen lamp in the range of 200–800 nm. White powder of BaSO4 was used as reference. During photocatalysis, 1.0 cm path length quartz cuvette and aqueous solution were used for background subtraction. Photoluminescence (PL) spectra were collected in (Fluoro max-4) Horiba Jobin Yvon Spectrofluorimeter. The samples were excited (λex) at λ = 315 nm. Emission slit widths were fixed to 5 nm each with a range 420 to 700 nm. Zeta potential analysis was performed using a Malvern Zetasizer, MAL 1040152 (Malvern Instruments, UK). All the measurements were carried out in room temperature under ambient condition.Structure and morphology
To determine the crystal structure and degree of lattice strain powder X-ray diffraction pattern was recorded using a Rigaku Mini Flex II diffractometer with Cu-Kα radiation at 25 °C. 2θ values were ranging from 10–80° and with scanning rate 2° per min. Room temperature Fourier transform infrared spectra of the powder samples (pellets in KBr, without moisture) were measured by using a Perkin Elmer 2000 infrared spectrometer in the range of 500–2000 cm−1. FESEM analysis was carried out using Nova NanoSem 450 operated from 0.5 kV to 30 kV. All the as-synthesized samples were analysed after coating with platinum metal. EDS measurements were performed using Bruker XFlash 6130, attached with FESEM instrument. Transmission electron microscopy imaging was carried out on a Bruker microscope operated at 200 kV. The samples were prepared onto 400-mesh carbon-coated copper grids by placing 3–4 drops of freshly prepared aqueous dilute solution and the grids were dried for overnight. During TEM measurement, all the samples were highly stable under the electron beam irradiation. TEM and HRTEM images were taken at different places to check the uniformity.Surface chemical properties
X-ray photoelectron spectroscopy (XPS) was performed using a commercial SPECS spectrometer (Germany), equipped with an Al-Kα X-ray source (1486.5 eV). High resolution XPS spectra were collected using pass energy of 40 eV with a step size of 0.05 eV. For all measurements emission current of the X-ray source was fixed at 15 mA and the UHV chamber base pressure was < 5 × 10−10 mbar. XPS samples were prepared by dropping 10.0 μL of the aqueous solution of g-C3N4/ZnO onto small pieces Si wafer and dried under dry nitrogen line. To compensate any kind of charging effect, the binding energy of Au 4f peaks were used as reference.To determine the surface areas of g-C3N4/ZnO heterostructures, N2 adsorption–desorption isotherms was measured with Autosorb iQ Station 1 (Quantachrome Instruments, version 3.01) at a bath temperature of 77.35 K.
Results and discussion
Synthesis and optical property
Synthesis of g-C3N4 and g-C3N4/ZnO heterostructures has been explained in the Experimental section. In brief, bulk g-C3N4 was synthesized using urea via poly-condensation reaction and different morphologies of ZnO were evolved on g-C3N4 surface via wet-chemical route. The resulting heterostructures were used in visible light photocatalysis reaction and showed a high stability and durability. The characterization, photocatalysis results and possible photodegradation mechanism of g-C3N4 and coupled g-C3N4/ZnO heterostructures are discussed in the following section.Fig. 1 shows UV-vis spectra of g-C3N4 and g-C3N4/ZnO heterostructures. g-C3N4 and g-C3N4/ZnO show strong absorption in visible light with absorption edge at ∼440 nm. The position of absorption edge remains unchanged for all the samples whereas g-C3N4/ZnO heterostructure shows an increased absorption as compared to the bare g-C3N4, which is due to the presence of ZnO nanostructure.23,31 The absorption intensity for both g-C3N4/ZnO heterostructure shapes are similar, which can be attributed to the UV-light absorption of ZnO. Hence, the morphological shape of various ZnO structures on g-C3N4 surface doesn't affect the absorption intensity within the visible region. Although, a sharp band at 369 nm for ZnO has been reported in the literature,33 g-C3N4/ZnO heterostructures do not show any distinct peak for ZnO within the UV-vis spectrum. The band gap (Eg) of g-C3N4 can be estimated from the plot of (αEp)2 vs. photon energy (Ep) using the extrapolated value of Ep at α = 0, following equation: αEp = k(Ep − Eg)1/2, where, α = absorption coefficient, Ep = discrete photon energy, Eg = band gap energy, and k = constant. The calculated Eg value for g-C3N4 is found to be ∼2.88 eV (Fig. 1 (inset)), which is consistent with previous reports.20
 | ||
| Fig. 1 UV-vis DRS for bulk g-C3N4, g-C3N4/ZnO (dumbbells), and g-C3N4/ZnO (cones). Where, all the powder samples were mixed with BaSO4 as reference. Here, all absorbance values are in arbitrary unit and each spectrum corresponds to different absorbance (not in relative scale). From the UV-vis spectrum of g-C3N4, inset shows (αEp)2 vs. Ep plot for band gap (Eg) calculation. | ||
In order to analyze the effect of ZnO and the fate of photogenerated electron–hole pairs on g-C3N4 surface, photoluminescence (PL) spectral analysis has been carried out. Fig. 2 shows the PL spectra of g-C3N4 and g-C3N4/ZnO heterostructures with an excitation wavelength of 315 nm. The intense emission peak is centred at around 441 nm for pure bulk g-C3N4, which could be assigned to the higher recombination rate of the photogenerated electrons and holes.12,32 In case of g-C3N4/ZnO heterostructures, red shifted emission peaks appear at 436 nm and 432 nm for dumbbell and cone structure, respectively. The decrease in emission peak intensity for g-C3N4/ZnO heterostructures is due to inhibited recombination of photogenerated electron–hole pairs in g-C3N4 and ZnO heterojunction, which further indicates a successful charge separation. However, PL study confirms about the increased lifetime of the photogenerated carrier in g-C3N4/ZnO heterostructures compared to bare g-C3N4, which is due to the type-II band alignment of ZnO and C3N4.
 | ||
| Fig. 2 Room temperature photoluminescence spectra of bulk-g-C3N4, g-C3N4/ZnO (dumbbells), and g-C3N4/ZnO (cones). For PL measurements all the powder samples were dispersed well in water using a sonicator with an excitation wavelength, λex = 315 nm. | ||
Structure and morphological characterization
The FTIR spectra for pure g-C3N4, ZnO, and composite g-C3N4/ZnO heterostructures are shown in ESI (Fig. S1†). In case of pure g-C3N4, the peak centred at 1636 cm−1 is attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration, while the peaks at 1240, 1318, and 1406 cm−1 represent the aromatic C–N stretching vibration modes of C3N4 ring. The out-of-plane bending modes of C–N heterocycles are represented by a band at ∼811 cm−1.20,21,30 From Fig. S1,† it is quite clear that the main characteristic peaks of g-C3N4 and ZnO appears in g-C3N4/ZnO heterostructures with a red shift, which indicates weaker bond strengths of C–N and CN within the g-C3N4/ZnO heterostructures. This phenomena suggest that the conjugated system of g-C3N4 is stretched and appears in a more widely conjugated system in g-C3N4/ZnO heterostructures.21 Therefore, FTIR spectra reveal the existence of g-C3N4, ZnO, and g-C3N4/ZnO, which are well matched with the reported stretching frequency values and further indicates an intense interaction between g-C3N4 and ZnO.20,21,24,30
N stretching vibration, while the peaks at 1240, 1318, and 1406 cm−1 represent the aromatic C–N stretching vibration modes of C3N4 ring. The out-of-plane bending modes of C–N heterocycles are represented by a band at ∼811 cm−1.20,21,30 From Fig. S1,† it is quite clear that the main characteristic peaks of g-C3N4 and ZnO appears in g-C3N4/ZnO heterostructures with a red shift, which indicates weaker bond strengths of C–N and CN within the g-C3N4/ZnO heterostructures. This phenomena suggest that the conjugated system of g-C3N4 is stretched and appears in a more widely conjugated system in g-C3N4/ZnO heterostructures.21 Therefore, FTIR spectra reveal the existence of g-C3N4, ZnO, and g-C3N4/ZnO, which are well matched with the reported stretching frequency values and further indicates an intense interaction between g-C3N4 and ZnO.20,21,24,30
In order to check the phase purity and structural property of the as-synthesized g-C3N4 and g-C3N4/ZnO composites, powder X-ray diffraction (PXRD) analysis has been performed, as shown in Fig. 3. The XRD pattern (inset of Fig. 3) of pure g-C3N4 reveals two distinct peaks at 2θ = 13.3° and 27.6°. The weaker peak at 13.3° corresponds to the presence of (100) crystalline plane, which is due to in-plane ordering of tri-s-triazine units. The relatively stronger peak at 27.6° is recognized as the reflection from (002) lattice plane which also resembles an interlayer spacing of d = 0.32 nm, the long-range interlayer stacking of the aromatic system of g-C3N4.12,13
 | ||
| Fig. 3 Powder X-ray diffraction patterns of bulk g-C3N4 (inset), g-C3N4/ZnO (dumbbells), and g-C3N4/ZnO (cones). During PXRD measurement, 2θ varies from 10–80° and the scanning rate was fixed at 2° per min. | ||
In case of g-C3N4/ZnO heterostructures, eight major reflection peaks for ZnO are observed at 2θ = 31.78°, 34.42°, 36.19°, 47.40°, 56.47°, 62.63°, 68.85°, and 76.71°, correspond to the (100), (002), (101), (102), (110), (103), (112), and (202) crystal planes of ZnO (Fig. 3). According to JCPDS card no. 89-1397, all the observed diffraction peaks match well with the wurtzite (HCP) structure of ZnO for both the cone and dumbbell shape morphologies.33 Apart from the eight major peaks for ZnO, another diffraction peak at 2θ = 27.6°, representing the (002) lattice plane of g-C3N4, also appears which confirms the existence of ZnO on g-C3N4 surface (Fig. 3). Absence of any impurity peak, originated from unreacted urea, non-hydrolyzed Zn2+ precursor salt, and Zn(OH)2, within the XRD pattern of g-C3N4 and g-C3N4/ZnO heterostructures also confirms the material's phase purity.
The morphological analysis of the as-synthesized ZnO has been studied using a field emission scanning electron microscopy (FESEM). Fig. 4a and b represent the low and high resolution FESEM images of ZnO structures, synthesized using HMT hydrolyzing agent. Fig. 4a shows an SEM image of randomly distributed dumbbell shaped ZnO structure on g-C3N4 surface maintaining the shape uniformity. The average length and breadth of ZnO dumbbells on g-C3N4 surface is about 1.76 μm and 0.76 μm, respectively, as can be seen in Fig. 4b. Fig. 4c and d exhibit FESEM images of cone shaped ZnO structures, where ammonia was utilized as hydrolyzing agent. The average length of the side of a cone is found to be about 0.51 μm (Fig. 4d), which is significantly shorter than the dumbbell shape. The presence of ZnO cone on g-C3N4 surface is aggregated even with an uniform shape. Pure bulk g-C3N4 also appears in aggregated morphology with many C3N4 sheets and layered structure (Fig. S2†). All these findings clearly suggest a random growth of ZnO on g-C3N4 surface, for both dumbbells and cones and results in different growth morphologies for HMT (dumbbell shape) and ammonia (cone shape) hydrolyzing agent.
 | ||
| Fig. 4 FESEM images of g-C3N4/ZnO dumbbells (a) at low resolution and (b) at high-resolution. FESEM images of g-C3N4/ZnO cones (c) at low resolution and (d) at high-resolution. | ||
The structures and shapes of ZnO dumbbells and cones on g-C3N4 surface have also been investigated with the help of TEM. TEM images of morphology dependent ZnO are shown in Fig. 5. Fig. 5a exhibits a TEM image of ZnO dumbbell on g-C3N4 surface, which was synthesized using the HMT. The length and breadth of a single dumbbell is found to be 1.73 μm and 0.78 μm (inset of Fig. 5a), respectively, which are complementary to our FESEM findings. In can also be noted that the size and shape of the ZnO dumbbells are quite uniform. High-resolution TEM image (Fig. 5b) of C3N4/ZnO heterostructure shows a fringe spacing value of 0.26 nm, which corresponds to the (002) crystal planes of ZnO. Hence, we can conclude that the growth of ZnO dumbbell on g-C3N4 surface occurred along (002) plane. TEM and HRTEM images of ZnO cones on g-C3N4 surface (synthesized by using ammonia) are shown in Fig. 5c and d. From the inset of Fig. 5c, the length of a cone can be estimated to 0.51 μm, well in line with earlier FESEM analysis. Fig. 5d shows clear lattice fringes of ZnO on g-C3N4 surface with fringe spacing of d = 0.26 nm, which can be attributed to the (002) lattice planes of the wurtzite ZnO crystal, and also complementary with our XRD results. Finally, TEM and HRTEM analysis also confirm the morphology dependent synthesis of ZnO using various hydrolyzing agents and their random distribution on g-C3N4 surface.
 | ||
| Fig. 5 (a) TEM and (b) HRTEM images of g-C3N4/ZnO dumbbell. (c) TEM and (d) HRTEM images of g-C3N4/ZnO cones. Inset of (a) and (c) shows a single dumbbell and cone at high-resolution. | ||
Selected area elemental mapping of g-C3N4/ZnO cone shaped heterostructures (Fig. S3†), has been performed by EDS technique which confirms the presence of C, N, Zn, and O elements. The line mapping of g-C3N4/ZnO heterostructure shows different lines for Zn, O, C, and N (Fig. S3a†) which clearly proves the random distribution of ZnO over the g-C3N4 surface.
N2 adsorption–desorption isotherms of the as-decorated dumbbell and cone shaped ZnO catalyst samples have been used to determine the surface areas of the catalysts, which further helps to quantify the adsorption property. The BET surface area values of 45.35 and 23.67 m2 g−1 have been observed for g-C3N4/ZnO dumbbells and cones, respectively. The larger surface area value of ZnO dumbbells confirms better adsorption property over the ZnO cones. The N2 adsorption–desorption isotherms of ZnO dumbbell and cone structures are presented in Fig. S4.†
Surface chemical analysis of ZnO on g-C3N4 surface
To investigate the surface chemical properties, relative surface coverages and the oxidation states of the dumbbell and the cone shaped ZnO structures synthesized on g-C3N4 surface, XPS technique has been employed as a characterizing tool. Successful formation of both types of ZnO shapes on g-C3N4 surface and relative comparison of their surface coverage as well as oxidation states are depicted in Fig. 6. Survey spectra for cone (ZnO-C) and dumbbell (ZnO-D) shaped structures on graphitic carbon nitride layers are shown in Fig. 6a. Both scans appear with strong existence of Zn, N, C, and O core level binding energy as well as Auger peaks, which further confirm the successful formation of ZnO on g-C3N4 surface. High resolution scans of Zn 2p core level spectra are shown in Fig. 6b. The binding energy positions for Zn 2P3/2 and Zn 2P1/2 are observed at around 1021.6 eV and 1044.7 eV, respectively, which are in good agreement with the earlier reported values for Zn(+2) oxidation state of ZnO material.24,28,30 However, stronger peak intensity in ZnO-C spectrum clearly suggests a significantly higher surface coverage for the cone-shaped structures on g-C3N4 as compared to that of the dumbbell shaped ZnO. Another comparison of the binding energy peak intensities for N1s line is also presented in Fig. 6c. In contrast to earlier Zn 2p lines, here, the N1s line appears stronger for ZnO-D materials which can be explained in terms of attenuation of the N1s photoelectrons emitted from the g-C3N4 surface, within the ZnO over layers for ZnO-C sample. This finding is very much complementary with our SEM results. In addition, it can be noted that there is a little relative shift in N1s binding energy peak position towards the higher energy for ZnO-C sample. This might be attributed to a relatively lower electron density of the N atoms within the ZnO-C. A similar kind of peak shift for O1s line in the opposite direction is also observed (Fig. 7), which can be correlated to a relatively higher electron density of the O atoms within the ZnO-C sample. Both these relative peak shifts (N1s and O1s) suggest that there are some possibilities for partial transfer of electrons from the N atom of g-C3N4 to the O atoms of ZnO. This assumption is also complementary with the higher electro-negativity of O compared to that of N. Therefore, we can conclude that a composite of g-C3N4/ZnO is obtained rather than a physical mixture of ZnO and g-C3N4 is formed within the samples.32 | ||
| Fig. 6 XPS spectra of (a) wide scan survey of cone (ZnO-C) and dumbbell (ZnO-D) shaped g-C3N4/ZnO structures. High resolution scans of (b) Zn 2p and (c) N1s core level photoelectrons. | ||
 | ||
| Fig. 7 High-resolution XPS spectra of g-C3N4/ZnO samples with various deconvolution components of C1s, N1s and O1s photoelectrons. (a–c) Cone morphology (ZnO-C) and (d–f) dumbbell morphology (ZnO-D). | ||
High-resolution XPS spectra of C1s, N1s and O1s binding energy peaks for both g-C3N4/ZnO samples are presented in Fig. 7. All spectral analysis with various de-convolution processes are performed using a linear background subtraction and a mixture of Lorentzian and Gauss fitting. Fig. 7a–c represent the corresponding spectra of ZnO-C sample whereas, Fig. 7d–f depict the spectral analysis for ZnO-D sample, respectively. C1s binding energy spectra of the ZnO-C and ZnO-D samples are shown in Fig. 7a and d, respectively. The deconvoluted components of C1s line appear in two main binding energy peaks positioned at about 287.7 and 284.6 eV. The peak cantered at 287.7 eV is identified as sp2 bonded carbon (N–CN), whereas the peak positioned at 284.6 eV corresponds to the graphitic carbon (C–C) of the materials and adventitious hydrocarbon from the instrument. Similar to earlier observation, Fig. 7b and e show the deconvoluted spectra of N1s for ZnO-C and ZnO-D samples, respectively. The asymmetrical shape of the observed N1s lines indicates chemically different N species within the g-C3N4. Two major components appear within the deconvoluted spectra are assigned to the sp2 hybridized nitrogen (C–NC) at lower binding energy and the sp3 hybridized tertiary nitrogen (C–N–C) at relatively higher binding energy. Additionally, a relative peak shift for ZnO-D samples toward the lower energy side can also be observed, which has already been discussed in earlier. The deconvolution of O1s spectra for both ZnO-C and ZnO-D samples can be seen in Fig. 7c and f, respectively. The oxygen species mainly appear in (−2) oxidation state within the crystalline ZnO matrix, with a minor fraction of surface hydroxides. Similar to N1s, a relative peak shift for O1s spectra in opposite direction originated from partial charge transfer between g-C3N4 and ZnO can also be noted. For better understanding and clarity, every details of all the deconvoluted spectra are summarized in Table 1.
| Sample | ZnO-C | ZnO-D | |||
|---|---|---|---|---|---|
| B.E. peaks | Sub-peaks | Peak position | Relative intensity | Peak position | Relative intensity |
| C1s | N–CN |
287.7 | 55585 |
287.7 | 86873 |
| C–C | 284.9 | 102606 |
284.8 | 109997 |
|
| N1s | C–NC |
398.9 | 59580 |
398.1 | 103713 |
| C–N–C | 400.4 | 34681 |
399.2 | 75326 |
|
| O1s | Zn–O | 530.9 | 199188 |
531.3 | 100847 |
| Zn–O–H | 532.7 | 19771 |
532.3 | 29034 |
|
Mechanism of formation of g-C3N4/ZnO heterostructures
The mechanism of the formation of bulk g-C3N4 is reported elsewhere.8–13 In brief, pyrolysis of urea takes place which afterwards results in a polymerized product. After successfully synthesizing the g-C3N4, it was washed with 0.1 M HNO3 to remove any kind of impurities. Dumbbell and cone structures of ZnO were formed on the surface of g-C3N4 by refluxing technique, using different hydrolyzing agent. At first, g-C3N4 powder was sonicated in water to obtained a well disperse layered structure of g-C3N4. Secondly, Zn2+ precursor salt and CTAB were added for the synthesis of g-C3N4/ZnO heterostructures. Finally, HMT and ammonia were used as hydrolyzing agents for the synthesis of dumbbell and cone shaped structures of ZnO, respectively.During the synthesis, CTAB was used as stabilizer and growth controlling agent for the growth of ZnO nanostructure on g-C3N4 surface. It is predicted that the surface of g-C3N4 is negatively charged (−24.6 ± 2.4 mV), which has also been confirmed from zeta-potential analysis. Therefore, Zn2+ precursor salt (ZnSO4·7H2O) can easily be adsorbed on negatively charged g-C3N4 surface and cationic surfactant (CTAB) can readily interact with adsorbed Zn2+ ions on the same surface. In addition, various hydrolyzing agents, such as HMT and ammonia were used to supply OH− ions in the solution, which is essential for the ZnO formation. Dumbbell shape (Fig. 4 and 5) of ZnO was obtained for a pH ∼ 7.5 of HMT, which acts as a rich source of OH− ions upon thermal degradation. Riley and co-workers reported that HMT acts as a source of OH− ion and its thermal decomposition produces ammonia and formaldehyde.34a Dutta and co-workers also reported the release of OH− ions upon thermal degradation of HMT.34b Whereas, for ZnO cone structures (Fig. 4 and 5), ammonia with pH ∼ 9 was used as a source of OH− ions, which spontaneously react with Zn2+ ions to form [Zn(OH)2] on g-C3N4 surface. Afterwards, the [Zn(OH)2] complex further reacts with excess OH− ions to form [Zn(OH)4]2− ions. During reflux, dehydration of [Zn(OH)4]2− takes place and finally gives rise to ZnO formation.
Apart from the OH− ion supply, as the rate of hydrolysis decreases with increasing pH and vice versa, HMT also acts as a buffer.34b Dutta and co-workers34b nicely represented the role of HMT for the growth of ZnO nanowire and nanorod. Due to the presence of a long chain polymer and a nonpolar chelating agent in HMT, it prefers to attach with the nonpolar facets of zincite crystal for epitaxial growth through a polar (001) face only.35a,b Therefore, instead of buffer like behaviour, HMT behaves more like a shape-inducing polymer surfactant. However, in this study, HMT was used for dual purposes. Firstly, a source of OH− ions and secondly a shape inducing polymer surfactant along with CTAB for the growth of ZnO dumbbells on the surface of g-C3N4. Finally, the surrounding of ZnO dumbbells on g-C3N4 surface is covered by the CTAB, which is confirmed by a positive surface charge (+5.14 ± 0.8 mV) from zeta potential analysis. There is no shape inducing agent for the synthesis of ZnO cones, the morphology was obtained solely due to the presence of cationic surfactant (CTAB) in the solution, a growth controlling agent. This phenomena was confirmed by a positive surface charge (+14.3 ± 1.2 mV) of g-C3N4/ZnO cone heterostructures from zeta potential analysis. Therefore, we can conclude that the CTAB and the available OH− ions from ammonia solution can favour the formation of ZnO cones on g-C3N4 surface. Due to relatively higher concentration of OH− ions from ammonia, the surface coverage of ZnO cones on g-C3N4 is found significantly more, whereas the coverage of ZnO dumbbells appears less with the HMT (Fig. 4 and 5). This relatively higher surface coverage of ZnO cones over dumbbell structures was also confirmed by XPS and SEM analysis. The overall formation mechanisms of ZnO in presence of both HMT and ammonia are given below.
| g-C3N4/Zn2+ + 2OH− → g-C3N4/Zn(OH)2 |
| g-C3N4/Zn(OH)2 + 2OH− → g-C3N4/Zn(OH)42− |
| g-C3N4/Zn(OH)42− → g-C3N4/ZnO + OH− + H2O |
Additionally, some control experiments were carried out to determine the role of CTAB, HMT, ammonia, for the formation of different ZnO morphologies on g-C3N4 substrate. At first, without any CTAB aggregated ZnO particles were obtained on the surface of g-C3N4 keeping all other conditions unaltered. FESEM image of aggregated ZnO particles is shown in ESI Fig. S5.† Secondly, in absence of any hydrolyzing agent (HMT or ammonia) it was impossible to grow any ZnO on the g-C3N4 surface. In an alternate approach, without using any C3N4 while keeping the CTAB and ammonia concentration unaltered, flower shaped ZnO structures were observed.33 However, other morphological shapes of ZnO such as nanorod, nanowire, nanotube etc. have been reported for HMT hydrolyser, without C3N4 substrate.36 Therefore, it can be concluded that presumably, C3N4 can provide many active centres for the initial nucleation of ZnO. This effect helps to grow ZnO randomly on C3N4 surface in spite of growing on a single centre which finally give rise to the flower, rod, wire etc. like morphology. Therefore, for the synthesis of ZnO dumbbell and cone structures on g-C3N4 surface, CTAB, HMT, ammonia, and g-C3N4 play very important roles. The overall formation of shape-dependent ZnO nanostructure on g-C3N4 surface is shown in Scheme 1.
 | ||
| Scheme 1 The overall mechanism for the formation of bulk g-C3N4, g-C3N4/ZnO heterostructure (dumbbell and cone), and aggregated ZnO on g-C3N4 surface. | ||
Photocatalysis
Photocatalytic dye degradation experiments with MB and phenol organic pollutants were performed under the illumination of visible light source with pure g-C3N4 as well as composite g-C3N4/ZnO dumbbell and cone heterostructure catalysts. During photocatalysis reaction, aqueous solution of 1 mL of 3 × 10−4 M MB, 0.4 mL of 5 × 10−3 M phenol and 5.0 mg of catalysts were used. The main characteristic peaks for MB and phenol at 664 nm and 270 nm were used to monitor the photocatalytic reaction. Tungsten bulb with a wavelength range of 300–1200 nm was used as a visible light source for the photocatalytic study. There was no degradation of MB and phenol in absence of visible light as well as without any catalyst. To check the dye adsorption on catalyst surface, the reaction mixture of MB solution and catalyst was agitated in the dark with continuous stirring, prior to any irradiation with visible light. In the dark, maximum adsorption of dye was found to be 20% (within 20 min) on the catalyst surface (Fig. S6†) and afterwards it remained constant till 140 min. Therefore, to stabilize the adsorption–desorption equilibrium, the reaction mixture (dye and catalyst) was stirred 20 min in dark condition prior to any use.It can be seen from Fig. S7† that the degradation of MB with bare g-C3N4 was reached ∼55% decomposition under visible light irradiation. The rate constant (kbare) value with bare g-C3N4 is observed 0.6 × 10−2 min−1 and showed pseudo-first order kinetics. Fig. 8a shows the degradation of the MB using dumbbell shape of g-C3N4/ZnO heterostructures. After 140 min, the reaction was almost over and ∼99% of dye degradation was achieved. Within Fig. 8b, At/A0 vs. time (min) has been plotted, which shows an exponential behaviour with time. The rate constant (kND) of 2.4 × 10−2 min−1 was determined from the slope of ln(At/A0) vs. time (min) plot (Fig. 8c), which is four times higher than that of bare g-C3N4. Fig. 8c also confirms pseudo-first-order kinetics of MB degradation using g-C3N4/ZnO dumbbell catalyst. Fig. S8a† shows the UV-vis absorption spectra of MB degradation using cone shaped g-C3N4/ZnO heterostructures. At the end of 140 min, ∼99% of the dye was already degraded. Similar to earlier result, the At/A0 vs. time (min) plot also appears in exponential nature (Fig. S8b†) and from the slope of ln(At/A0) vs. time (min) plot (Fig. S8c†), the rate constant (kNC) was calculated to be 1.8 × 10−2 min−1, which is three times higher than that of bare g-C3N4. Pseudo-first-order kinetics of MB degradation was also observed with cone structures of ZnO. After the completion of 1st cycle, the catalyst was removed from the reaction mixture and washed for the re-use purpose.
 | ||
| Fig. 8 UV-vis spectra of (a) visible light degradation of MB in presence of g-C3N4/ZnO (dumbbells), (b) At/A0 vs. time (min) plot and (c) ln(At/A0) vs. time (min) plot. During the photocatalysis measurements, reaction mixture was stirred continuously. Conditions: [MB] = 3 × 10−5 M and amount of catalyst = 5.0 mg. In all cases water was used as reference solution. | ||
Furthermore, to prove the visible light activity of g-C3N4/ZnO (dumbbell and cone) heterostructures, a pure UV light active material phenol was used for degradation reaction. The UV-vis spectra of phenol degradation using dumbbell and cone shaped ZnO heterostructures are shown in ESI Fig. S9a and b,† respectively. It can be seen from Fig. S9a and b† that the complete phenol degradation was reached under a visible light irradiation in 150 and 170 min for dumbbell and cone structure of ZnO, respectively. Whereas, 300 min is required for complete degradation of the phenol with bare g-C3N4 catalyst. The rate constant values for g-C3N4/ZnO dumbbell and cone heterostructure are found 2.0 × 10−2 min−1 and 1.5 × 10−2 min−1, respectively, which is significantly higher than that of pure g-C3N4. Hence, phenol degradation result also confirms the higher photo-activity of the dumbbell and cone structure of ZnO on g-C3N4 surface as compared to the bare g-C3N4 catalyst.
Fig. 9 shows the comparative study of MB dye degradation using ZnO, bare g-C3N4, and g-C3N4/ZnO (dumbbell and cone) heterostructure catalysts under visible light irradiation. Pure ZnO nanoparticles are UV light active and does not have any role on MB dye degradation under visible light illumination.33 Bare g-C3N4 is known as a visible light active material and electron–hole pairs are generated after irradiation with visible light. It can be seen from Fig. 9 that the degradation of MB occurred with bare g-C3N4 with a slowest rate (kbare = 0.6 × 10−2 min−1), which is due to the fast recombination of the photogenerated charged species (e− and h+). Therefore, to suppress this problem of recombination, a heterojunction was created by modifying the surface of g-C3N4 with dumbbell and cone structures of ZnO. In case of heterostructures, the photo-activity is higher (kND = 2.4 × 10−2 min−1 and kNC = 1.8 × 10−2 min−1) due to the decreased recombination of e− and h+. This diminished recombination of e−–h+ pairs has also been proved from the decreased intensity in photoluminescence spectra (Fig. 2) of g-C3N4/ZnO heterostructures as compared to bare g-C3N4. Sun and co-workers reported 3 times better activity in visible light dye degradation with g-C3N4–ZnO spherical particles as compared to bare g-C3N4 for methyl orange and p-nitrophenol degradation.20 Kumar and co-workers also reported 5 times higher rate constant value for N-doped ZnO nanoplates than bare g-C3N4 nanosheets.30 Li and co-workers reported 3.8 times higher rate constant value with g-C3N4/ZnO composite than the pure g-C3N4, during rhodamine B dye degradation.37 However, both dumbbell and cone structures of ZnO on g-C3N4 surface shows 3–4 times higher activity in MB and phenol dye degradation than bare g-C3N4, under illumination with the visible light and the rate constants order is found as kND > kNC > kbare. The higher rate constant with g-C3N4/ZnO dumbbell structures is mainly due to the larger surface area of dumbbells (45.35 m2 g−1) as compared to that of cones (23.67 m2 g−1). Larger surface area of the catalyst particles absorb more organic dye molecules, which further confirms the faster dye degradation rate of ZnO dumbbells than cone structures. Therefore, the as-obtained morphologies of ZnO in g-C3N4/ZnO heterostructures show better photo-activity than other reported ZnO morphologies.
 | ||
| Fig. 9 Comparative studies of degradation of MB dye in presence of ZnO, bulk g-C3N4, g-C3N4/ZnO (dumbbells), and g-C3N4/ZnO (cones) under visible light. | ||
Mechanism for enhanced photo-activity with g-C3N4/ZnO heterostructure
In case of heterogeneous photocatalysis, two important parameters are the adsorption of dye on catalyst surface and the degradation of dye with the help of photogenerated electron–hole separation. In this study, dumbbell and cone morphologies of g-C3N4/ZnO heterostructures and negative surface charge of g-C3N4 enhance the adsorption process of dye on its surface. The efficient photo-activity of g-C3N4/ZnO heterojunctions can be explained in terms of the alignment of the valence band (VB) and the conduction band (CB) positions of both g-C3N4 and ZnO separately. Both the g-C3N4 and ZnO are examples of type-II semiconductor, which can be verified by potential energy level calculations as shown in the following equation:38–42| EVB = χ – Ec + 0.5Eg |
| ECB = EVB − Eg |
The value of χ for ZnO is 5.79 eV and the band gap energy (Eg) of the as-synthesized ZnO material is 3.12 eV. The EVB of ZnO was calculated to be +2.85 eV. Hence, the ECB of ZnO can be estimated to −0.27 eV. Based on the reported data, the CB and the VB edge potential values of g-C3N4 are −1.1 eV and +1.6 eV, respectively.38,41 All the potential energy values for both ZnO and g-C3N4 are shown in Scheme 2. It can be seen from Scheme 2 that the CB potential energy or LUMO for g-C3N4 (ECB = −1.1 eV) is lower than the ZnO (ECB = −0.27 eV). Hence, the excited e− from the CB of g-C3N4 can easily inject to the CB of ZnO upon irradiation with visible light.21,30–32,38 Therefore, the potential energy match in the heterojunction of g-C3N4/ZnO is a driving force for the transportation of photogenerated e− from the CB of g-C3N4 towards the CB of ZnO, which effectively reduces the recombination of e−–h+ pairs.32,38
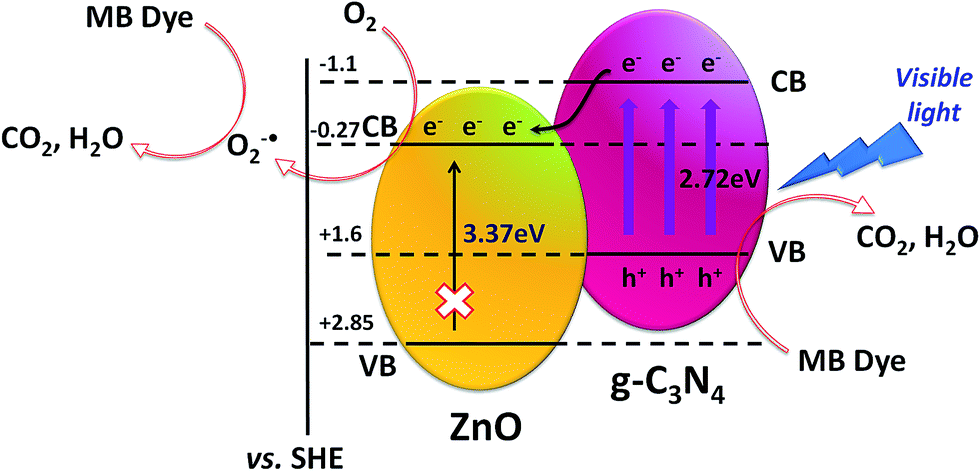 | ||
| Scheme 2 The VB and CB energy levels and the mechanism of charge transfer in g-C3N4/ZnO heterostructure under visible light irradiation during organic pollutant degradation. | ||
Upon irradiation of visible light on g-C3N4/ZnO heterostructures, the e− on g-C3N4 can easily move from VB to CB (eCB−) leaving behind h+ on VB (hVB+) of g-C3N4. Now, the excited electrons may easily transfer from the CB of g-C3N4 to the CB of ZnO. Afterwards, the electrons on the CB of ZnO react with O2 and generate superoxide radicals (˙O2−), which further generate hydroxyl radicals (˙OH).21,30–33,43 These ˙OH radicals are responsible for the photodecomposition of MB and phenol. On the other hand, h+ in VB of g-C3N4 is also responsible for the generation of ˙OH radicals from OH− ions, which are the active species in dye degradation.20,21,33,43 As ZnO is an UV light active material, photo-excitation of it is not possible under visible light irradiation. The overall electronic transition and dye degradation reaction in g-C3N4/ZnO heterostructures are shown below:

It can be seen from the above reactions that the species like e−, ˙O2−, ˙OH, and h+ are directly involved in the photocatalytic process.44,45 Therefore, to confirm the role of electrons and holes in visible light photocatalysis, scavenging experiment has been carried out with t-BuOH and ammonium oxalate (AO) for MB degradation, using g-C3N4/ZnO heterostructure catalyst. t-BuOH was used as electron/radical scavenger and AO as a hole scavenger.30,33 For scavenging experiments, 1 mL of t-BuOH and 0.2 g of AO were used, keeping the other reaction parameters unaltered, to trap the electron/radical and holes, respectively. Incorporation of t-BuOH in the reaction mixture prevented the presence of e− and the formation of ˙O2− and ˙OH radicals, which largely affected the visible light dye degradation and results in ∼27% degradation. On the other hand, h+ was promptly scavenged with the addition of AO and ∼88% degradation was achieved. Therefore, it can be concluded that % degradation of dye is not greatly affected due to the h+ scavenger. Altogether, the formation of ˙O2− and ˙OH radicals play a major role in photocatalysis under the visible light. This scavenging experiment findings are also supported by earlier reported values.21,30,31
Reusability of g-C3N4/ZnO catalyst
Reusability of g-C3N4/ZnO catalyst has been tested in MB degradation reaction, under the irradiation of visible light source. Fig. 10a shows the reusability histogram using ZnO dumbbell for MB degradation. After the 1st cycle, catalyst particles were collected and washed thoroughly with water for further use. Using same condition, the experiments were repeated thrice and the % of degradation results show 98%, 94%, and 92% for 1st, 2nd, and 3rd cycles, respectively. This degradation result confirms that the catalysts remain chemically stable even after several recycles. Similar reusability test was also conducted with ZnO cones and the % degradation demonstrate 98%, 92%, and 90% for 1st, 2nd, and 3rd cycles, respectively (Fig. 10b). After the 3rd cycle of photocatalysis reaction, both (dumbbell and cone) morphologies of ZnO in g-C3N4/ZnO heterostructures were checked with the help of FESEM which also confirmed the morphological stability of the ZnO structures (Fig. S10†). | ||
| Fig. 10 Histogram between % degradation vs. number of cycle for (a) g-C3N4/ZnO (dumbbell) and (b) g-C3N4/ZnO (cone) catalyst after 140 min exposure (for each cycle) under a visible light source. The catalyst particles were centrifuged and washed well before reuse. | ||
Structural stability of the ZnO catalysts after several reuse was also verified using XRD analysis. Fig. S11† represents eight major reflection peaks of ZnO in both dumbbell and cone shaped particles appear at 31.78°, 34.42°, 36.19°, 47.40°, 56.47°, 62.63°, 68.85°, and 76.71° confirm the presence of (100), (002), (101), (102), (110), (103), (112), and (202) crystal planes of ZnO, respectively. The other 2θ value at 27.6° is due to the (002) plane of g-C3N4. These findings are very much in line with earlier XRD data of the ZnO structures before any dye degradation. Therefore, it can be concluded that the structural purity and phase stability of dumbbell and cone shaped ZnO remain unchanged even after several cycles of the degradation process.
Conclusion
In conclusion, we have successfully developed a simple and facile wet-chemical approach for the synthesis of morphology-dependent ZnO on g-C3N4 surface. Dumbbell and cone structures of ZnO were synthesized using HMT and ammonia as hydrolyzing agents. As characterizing techniques, FESEM, XRD, TEM, XPS, and EDS were used to verify the formation of dumbbell and cone morphology of ZnO on g-C3N4 surface and phase purity as well as investigate their structural and chemical properties. On the other hand, UV-vis, PL, and FTIR were used to confirm the existence and increased lifetime of the photogenerated carriers in g-C3N4/ZnO heterostructures. The shape-dependent mechanism using HMT and ammonia hydrolyzing agent was discussed thoroughly. The as-prepared heterostructures were used in organic pollutants degradation under visible light irradiation which show a higher visible light photo-activity for g-C3N4/ZnO heterostructures than bare ZnO particles and g-C3N4. Moreover, a better photo-activity was observed with the g-C3N4/ZnO dumbbells than the g-C3N4/ZnO cone structure. This simple, environmental friendly and the unique hybrid structures of g-C3N4/ZnO make the composite material a greater prospective in the field of energy and environment. Finally, this result provides a new visible light driven environmentally benign catalyst with high stability and shows a new pathway for fabricating an efficient heterostructure photocatalyst.Acknowledgements
SP gratefully acknowledges the financial support from Department of Science and Technology (DST) Science and Engineering Research Board (SERB) Fast track (SB/FT/CS-042/2012) grant and BITS additional competitive research grant, No. PLN/AD/2014-15/June/04. PF gratefully acknowledges the financial support from UGC-BSR and Dr A. Kumar, UOR, Jaipur for helpful suggestion. We are also thankful to the University Grants Commission (UGC) special assistance program (F.540/14/DRS/2007 (SAP-I)), DST-FIST program, Government of India. Our sincere thank to Dr Mrinmoyee Basu, National Taiwan University for her valuable discussion. The instrumental support for TEM and FESEM measurements from Mr M. Reza of the Material Research Center (MRC), MNIT Jaipur is highly acknowledged. We also thank the Department of Physics, BITS Pilani for assistance with powder X-ray diffraction studies (DST-FIST sponsored).References
-
(a) Y. Zhang, Q. Pan, G. Chai, M. Liang, G. Dong, Q. Zhang and J. Qiu, Sci. Rep., 2013, 3, 1943 Search PubMed
; (b) A. Y. Liu and M. L. Cohen, Science, 1989, 245, 841 CAS
-
(a) Z. Ding, X. Chen, M. Antonietti and X. Wang, ChemSusChem, 2011, 4, 274–281 CAS
-
(a) J. Yang, X. Wu, X. Li, Y. Liu, M. Gao, X. Liu, L. Kong and S. Yang, Appl. Phys. A, 2011, 105, 161 CrossRef CAS
- Y. Iwano, T. Kittaka, H. Tabuchi, M. Soukawa, S. Kunitsugu, K. Takarabe and K. Itoh, Jpn. J. Appl. Phys., 2008, 47, 7842 CrossRef CAS
- J. Hong, X. Xia, Y. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006 RSC
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18 CrossRef CAS PubMed
- Y. Li, J. Zhang, Q. Wang, Y. Jin, D. Huang, Q. Cui and G. Zou, J. Phys. Chem. B, 2010, 114, 9429 CrossRef CAS PubMed
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS PubMed
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed
- X. C. Wang, X. F. Chen, A. Thomas, X. Z. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609 CrossRef CAS PubMed
- Y. Wang, H. Li, J. Yao, X. C. Wang and M. Antonietti, Chem. Sci., 2011, 2, 446 RSC
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171 RSC
- J. Liu, T. Zhang, Z. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398 RSC
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Muller, R. Schlogl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893 RSC
- X. Zhang, H. Wang, H. Wang, Q. Zhang, J. Xie, Y. Tian, J. Wang and Y. Xie, Adv. Mater., 2014, 26, 4438 CrossRef CAS PubMed
- H. Yan, Chem. Commun., 2012, 48, 3430 RSC
- K. Takanabe, K. Kamata, X. Wang, M. Antonietti, J. Kubota and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 13020 RSC
- Y. Di, X. C. Wang, A. Thomas and M. Antonietti, ChemCatChem, 2010, 2, 834 CrossRef CAS PubMed
-
(a) S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894 CrossRef CAS PubMed
- J. Sun, Y. Yuan, L. Qiu, X. Jiang, A. Xie, Y. Shen and J. Zhu, Dalton Trans., 2012, 41, 6756 RSC
- Y. Wang, R. Shi, J. Lin and Y. Zhu, Energy Environ. Sci., 2011, 4, 2922 CAS
-
(a) J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2010, 49, 441 CrossRef CAS PubMed
- X. C. Wang, K. Maeda, X. F. Chen, K. Takanabe and K. Domen, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS PubMed
- W.-K. Jo and N. C. S. Selvam, J. Hazard. Mater., 2015, 299, 462 CrossRef CAS PubMed
- Y. J. Zhang, T. Mori, J. H. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294 CrossRef CAS PubMed
- G. Liu, P. Niu, C. H. Sun, S. C. Smith, Z. G. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642 CrossRef CAS PubMed
- Y. Wang, Y. Di, M. Antonietti, H. R. Li, X. F. Chen and X. C. Wang, Chem. Mater., 2010, 22, 5119 CrossRef CAS
- Y.-P. Zhu, M. Li, Y.-L. Liu, T.-Z. Ren and Z.-Y. Yuan, J. Phys. Chem. C, 2014, 118, 10963 CAS
- H. Yan and H. Yang, J. Alloys Compd., 2011, 509, L26 CrossRef CAS PubMed
- S. Kumar, A. Baruah, S. Tonda, B. Kumar, V. Shanker and B. Sreedhar, Nanoscale, 2014, 6, 4830 RSC
- W. Liu, M. Wang, C. Xu and S. Chen, Chem. Eng. J., 2012, 209, 386 CrossRef CAS PubMed
- J. Wang, F.-Y. Su and W.-D. Zhang, J. Solid State Electrochem., 2014, 18, 2921 CrossRef CAS
- P. Fageria, S. Gangopadhyay and S. Pande, RSC Adv., 2014, 4, 24962 RSC
-
(a) M. N. R. Ashfold, R. P. Doherty, N. G. N. Angwafor, D. J. Riley and Y. Sun, Thin Solid Films, 2007, 515, 8679 CrossRef CAS PubMed
-
(a) K. Govender, D. S. Boyle, P. B. Kenway and P. O'Brien, J. Mater. Chem., 2004, 14, 2575 RSC
-
(a) L. Vayssieres, K. Keis, S. E. Lindquist and A. Hagfeldt, J. Phys. Chem. B, 2001, 105, 3350 CrossRef CAS
- X. Li, M. Li, J. Yang, X. Li, T. Hu, J. Wang, Y. Sui, X. Wu and L. Kong, J. Phys. Chem. Solids, 2014, 75, 441 CrossRef CAS PubMed
- J. Zhou, M. Zhang and Y. Zhu, Phys. Chem. Chem. Phys., 2014, 16, 17627 RSC
- X.-J. Wang, W.-Y. Yang, F.-T. Li, Y.-B. Xue, R.-H. Liu and Y.-J. Hao, Ind. Eng. Chem. Res., 2013, 52, 17140 CrossRef CAS
-
(a) Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543 CAS
- Y. Wang, Z. Wang, S. Muhammad and J. He, CrystEngComm, 2012, 14, 5065 RSC
- X. Lin, X. Guo, W. Shi, L. Zhao, Y. Yan and Q. Wang, J. Alloys Compd., 2015, 635, 256 CrossRef CAS PubMed
- X. Lin, X. Guoa, W. Shi, F. Guo, H. Zhai, Y. Yan and Q. Wang, Catal. Commun., 2015, 66–67 Search PubMed
- X. Lin, X. Guo, D. Liu, Q. Wang, H. Zhai and L. Chang, Mater. Res. Bull., 2015, 63, 72 CrossRef CAS PubMed
- F. Guo, W. Shi, X. Lin, X. Yan, Y. Guo and G. Che, Sep. Purif. Technol., 2015, 141, 246 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12463h |
| This journal is © The Royal Society of Chemistry 2015 |