
Investigation of quinoline-4-carboxylic acid as a highly potent scaffold for the development of alkaline phosphatase inhibitors: synthesis, SAR analysis and molecular modelling studies†
Imtiaz Khan‡§
a,
Syed Jawad Ali Shah‡b,
Syeda Abida Ejaz‡b,
Aliya Ibrar‡a,
Shahid Hameed*a,
Joanna Leckacd,
Jose Luis Milláne,
Jean Sévignycd and
Jamshed Iqbal*b
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad-45320, Pakistan. E-mail: shameed@qau.edu.pk; Fax: +92 51 9064 2241; Tel: +92 51 9064 2133
bCentre for Advanced Drug Research, COMSATS Institute of Information Technology, Abbottabad-22060, Pakistan. E-mail: drjamshed@ciit.net.pk; Fax: +92-992-383441; Tel: +92-992-383591/+92-992-383596
cDépartement de microbiologie-infectiologie et d'immunologie, Faculté de Médecine, Université Laval, Québec, QC, Canada
dCentre de Recherche du CHU de Québec – Université Laval, Québec, QC, Canada
eSanford Children's Health Research Center, Sanford-Burnham Medical Research Institute, La Jolla, CA 92037, USA
First published on 24th July 2015
Abstract
The role played by organic chemistry in the pharmaceutical industry continues to be one of the main drives in the drug discovery process. More than ever, the industry demands from organic chemists the development of small molecules, which could be a rich source of biological potential. In this context, a diverse range of quinoline-4-carboxylic acid derivatives has been synthesized and evaluated as potent inhibitors of alkaline phosphatases. The structural build-up of the synthesized compounds was based on the spectro-analytical data. Most of the tested compounds showed remarkable inhibition of human tissue-nonspecific alkaline phosphatase (h-TNAP), tissue specific human intestinal alkaline phosphatase (h-IAP) and human placental alkaline phosphatase (h-PLAP). Among them, 3j was identified as a potent inhibitor of h-TNAP with an IC50 value of 22 ± 1 nM, whereas, 3e emerged as a lead candidate against h-IAP and h-PLAP with IC50 values of 34 ± 10 and 82 ± 10 nM, respectively. 3a was a potent inhibitor of human germ cell alkaline phosphatase (h-GCAP) with an IC50 value of 150 ± 70 nM. The putative binding sites of the most potent inhibitors were inferred from molecular docking simulations using homology models based on the h-PLAP structure.
Introduction
Alkaline phosphatases (APs, EC 3.1.3.1) are dimeric enzymes with the ability to dephosphorylate phosphoesters of low molecular mass,1 and perform phosphotransferase,2 and protein phosphatase reactions.3 This isozyme family is comprised of two groups: the tissue-specific alkaline phosphatases which include the intestinal (IAP), placental (PLAP) and germ cell (GCAP) isozymes and the tissue-nonspecific alkaline phosphatase (TNAP), with, each isozyme encoded by different genes.4,5 Tissue-specific and tissue-nonspecific isozymes can be discriminated by their amino acid sequences and also by their inhibition by inhibitors. TNAP is inhibited by levamisole, histidine and homoarginine but not by L-phenylalanyl-glycyl-glycine (Phe-Gly-Gly), while the reverse is true for the tissue-specific isozymes.6,7Though the expression of TNAP is ubiquitous, it is highly expressed in the developing neural tube, kidney, liver and mineralizing tissues such as cartilage and bone. Its function is intimately associated with mineralization of skeletal and dental tissues, as deficiency in TNAP function in humans and mice leads to a heritable form of rickets/osteomalacia known as hypophosphatasia.8 The major function of TNAP in bone tissue is the degradation of extracellular inorganic pyrophosphate (PPi), a potent inhibitor of calcification, to enable the normal mineralization of skeletal and dental tissues.9 Thus, inhibitors of TNAP have the potential to be used as a drug to treat disorders of pyrophosphate metabolism such as in generalized arterial calcification of infancy and related rare genetic diseases as well as in chronic kidney disease.9,10
On the other hand, placental alkaline phosphatase (PLAP) is expressed in large amounts in the syncytiotrophoblast cells of the placenta from about the eighth week of gestation throughout pregnancy.11 PLAP was one of the first enzymes recognized as an oncofetal protein expressed in a variety of cancers.12,13 The germ cells alkaline phosphatase (GCAP) is also expressed in the placenta, although at 1/50 the level of PLAP,14 but it is expressed in testicular germ cell tumors of the testis, particularly seminomas.15,16 PLAP and GCAP are often co-expressed in ovarian cancers,17 and it has been suggested that transformation from normal to malignant trophoblast might be associated with a switch from PLAP to GCAP expression.18
Traditionally, levamisole and theophylline have been the only available inhibitors of TNAP with Ki values of 16 and 82 μM, respectively (Fig. 1).19 Recently, however, several groups have screened for and optimized small molecules as efficient and selective inhibitors of AP isozymes.9,10,20
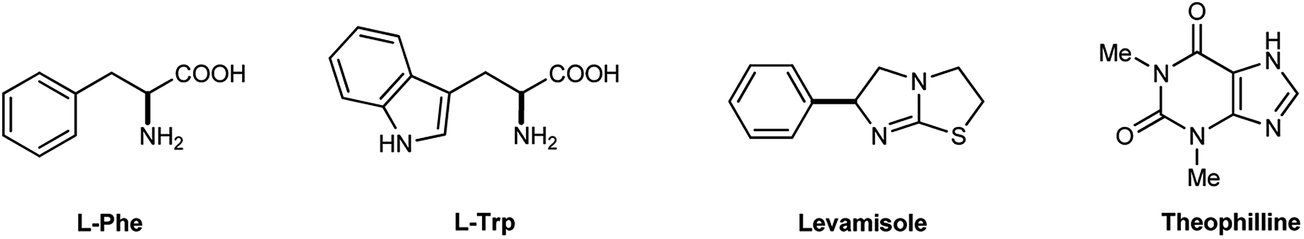 | ||
| Fig. 1 Selected examples of AP inhibitors. | ||
Nitrogen is a key component of many natural products and drug molecules. It has been estimated that among all natural products, the average number of nitrogen atoms per molecule is 0.7, while for medicinal drugs, this number rises to 3.0. As an important class of nitrogen-containing heterocycles, quinoline is one of the ubiquitous and privileged structural motifs that occur in bioactive natural products and pharmaceutically active therapeutic agents.21 Quinoline skeleton is also one of the key building elements for a large number of natural and synthetic heterocycles which are associated with a range of biological effects such as antimicrobial,22 anticancer,23 antimalarial,24 and antiviral activities.25 In addition, some natural, semisynthetic and synthetic bioactive molecules based on a quinoline scaffold have been reported to possess multidrug resistance MDR reversal activity when combined with anticancer drug.26 Numerous quinoline derivatives also display anti-HIV, antiasthmatic, antitumor, P-selectin antagonism, and antioxidant potential.27
Keeping in view the literature findings demonstrating the great interest in the research of new bioactive heterocycles and our continued interest in the development of potential AP inhibitors,28 we have synthesized quinoline-4-carboxylic acid derivatives as a new class of APs inhibitors with enhanced inhibitory potential against human TNAP, IAP, and PLAP. Homology modelling and docking studies were also performed to identify the putative binding modes of the top ranked inhibitors. The results of this study are presented in this paper.
Results and discussion
Synthesis
Quinoline-4-carboxylic acid derivatives (3a–j) were synthesized by the reaction of 5-chloro-isatin 1 with corresponding aryl substituted acetophenones (2a–j) in the presence of potassium hydroxide followed by acidification (Scheme 1).29 Structural modifications were validated by using a range of substituents (electron-rich and electron-poor) on the aryl ring of acetophenones delivering corresponding condensed products in good yields. Substituents were also tested on variable positions of acetophenones. | ||
| Scheme 1 Synthesis of quinoline-4-carboxylic acids (3a–j). | ||
Spectroscopic characterization
The formation of compounds 3a–j was confirmed through 1H NMR spectroscopy where 3a–j exhibited characteristic signals for carboxylic acid (–COOH) group in the range of 14.50–14.14 ppm along with additional aromatic protons appearing at appropriate chemical shift values. The disappearance of methyl protons of acetophenone also confirmed smooth cyclization. In 13C NMR spectra, distinctive signals in the range of 175.34–155.35 ppm were attributed to the carbonyl carbon of carboxylic acid functional group. The signals for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N functional group were also found in the appropriate region. The disappearance of the resonances for the carbonyls (ketone and amide) also supported the formation of quinoline-4-carboxylic acids (3a–j). The purity of the synthesized compounds was ascertained by elemental analysis.
N functional group were also found in the appropriate region. The disappearance of the resonances for the carbonyls (ketone and amide) also supported the formation of quinoline-4-carboxylic acids (3a–j). The purity of the synthesized compounds was ascertained by elemental analysis.
Biological activity
| Entry | Substituent (R) | Alkaline phosphatase inhibition | |||
|---|---|---|---|---|---|
| h-TNAP | h-IAP | h-PLAP | h-GCAP | ||
| IC50 ± SEM (μM)/% inhibitiona | |||||
| a %inhibition measured at 0.02 mM concentration of inhibitor (experiments performed in triplicate). | |||||
| 3a | 2-Br | 0.42 ± 0.02 | 0.075 ± 0.001 | 0.32 ± 0.01 | 0.15 ± 0.07 |
| 3b | 3-Br | 0.11 ± 0.07 | 0.14 ± 0.01 | 0.19 ± 0.02 | 41a |
| 3c | 3-F | 0.23 ± 0.06 | 43.5a | 0.19 ± 0.03 | 45a |
| 3d | 3-Me | 1.1 ± 0.2 | 36.2a | 0.28 ± 0.04 | 47a |
| 3e | 4-OMe | 0.36 ± 0.01 | 0.03 ± 0.01 | 0.08 ± 0.01 | 29a |
| 3f | 4-I | 1.91 ± 0.97 | 0.21 ± 0.01 | 1.07 ± 0.21 | 31a |
| 3g | 3-F–4-OMe | 1.39 ± 0.17 | 0.49 ± 0.17 | 0.74 ± 0.12 | 46a |
| 3h | 3-I–4-OMe | 0.13 ± 0.02 | 0.84 ± 0.13 | 0.21 ± 0.03 | 40a |
| 3i | 2,4-diOMe | 0.12 ± 0.01 | 0.35 ± 0.01 | 0.39 ± 0.01 | 0.29 ± 0.11 |
| 3j | 4-OH–3-OMe | 0.022 ± 0.001 | 0.27 ± 0.04 | 0.65 ± 0.09 | 48a |
| Levamisole | 25.2 ± 1.9 | — | 120 ± 2.5 | — | |
| L-Phenylalanine | — | 100 ± 3 | — | 301 ± 5 | |
Interestingly, most of the screened compounds were highly active and inhibited these APs much more efficiently than levamisole and L-phenylalanine. Among the tested compounds, 3j was identified as a potent inhibitor of TNAP with an IC50 value of 22 ± 1 nM, whereas, 3e emerged as a lead inhibitor of IAP and PLAP with an IC50 value of 0.034 ± 0.01 and 0.082 ± 0.01 μM, respectively. Compound 3a was identified as a potent inhibitor of GCAP with an IC50 value of 0.15 ± 0.07 μM.
The synthesized compounds 3a–j also showed remarkable inhibitory potency of IAP. The compound 3e incorporating para-methoxy substituent displayed the highest inhibitory activity with an IC50 value of 0.03 ± 0.01 μM as compared to the positive control L-phenylalanine (IC50 = 100 ± 3 μM). This inhibitory activity strength is 3000-fold higher as compared to L-phenylalanine which is presumably due to the interactions of amino acid residues with the methoxyl oxygen atom in addition to the other hydrogen bonding contacts making the inhibitor 3e stabilized in the active pocket of the target. Interestingly, incorporation of a 2-bromo substituent also produced good inhibition character with an IC50 value of 0.075 ± 0.001 μM which inhibit enzymatic activity ∼1335-fold more potently than standard drug. On moving bromo substituent from ortho- to meta-position produced diminished results as compared to compounds 3a and 3e, but still this potential is ∼700-fold higher as compared to L-phenylalanine. Surprisingly, the replacement of bromo substituent with fluoro group or an electron-donating methyl group led to a significant loss of inhibitory activity (compounds 3c and 3d; 43% and 36% inhibition, respectively). Other substitutions such as disubstitution of the aromatic ring also appeared to be tolerated possessing significant inhibitory activity (IC50 = 0.27–0.84 μM). This observation is mirrored in analogues 3g–3j, incorporating a combination of electron-rich and electron-deficient functional groups.
Similarly, compounds 3a–j were also tested for their biological activity against PLAP where compound 3e was identified as a strong inhibitor with an IC50 value of 0.08 ± 0.01 μM as compared to levamisole (IC50 = 120 ± 3 μM). This enhanced inhibitory ability of the compound was ∼1463-fold stronger as compared to L-phenylalanine (positive) control indicating that the methoxy substituent is necessary for the increased inhibitory activity when placed at para-position of the aryl ring. Aromatic ring attached to the quinoline core at 2-position also tolerated bromo, fluoro and methyl groups at ortho- and meta-position and the inhibitory activity results were promising (IC50 = 0.19–0.32 μM). We also investigated the effect of the double substitution on the aromatic ring, preparing the 3-F–4-OMe– (3g), 3-I–4-OMe– (3h), 3,4-diOMe– (3i) and 4-OH-3-OMe– (3j) analogues. The results revealed that compounds are potent inhibitors of PLAP with strong inhibitory activity depicting IC50 values in the range of 0.2–0.7 μM. This inhibition impact was much higher as compared to the levamisole (IC50 = 120 ± 3 μM).
Furthermore, when the same compound library was screened against GCAP, only compounds 3a and 3i showed extremely high inhibitory potential with an IC50 value of 0.15 ± 0.07 and 0.29 ± 0.11 μM, respectively. This inhibition data depicts ∼2000- and 1037-fold higher inhibition, respectively for 3a and 3i, as compared to the L-phenylalanine (IC50 = 301 ± 5 μM). The rest of the analogues showed less than 50% inhibition.
Taken together, the activity results suggest that the variation of the substituent position as well as their nature significantly affect the inhibition potential against TNAP, IAP, and PLAP. These studies also clearly demonstrated that the synthesized compounds have a significant potential to be developed as alkaline phosphatase inhibitors.
 | ||
| Fig. 2 Double-reciprocal plots of the inhibition kinetics of human tissue non-specific alkaline phosphatase (TNAP) indicating uncompetitive inhibition by compound 3j. Changes in the initial velocities of the reaction were measured at different concentrations of the inhibitor 3j using substrate CDP-Star (disodium 2-chloro-5-(4-methoxyspiro[1,2-dioxetane-3,2′-(5-chloro)tricyclo[3.3.1.13.7]decan]-4-yl)-1-phenyl phosphate). | ||
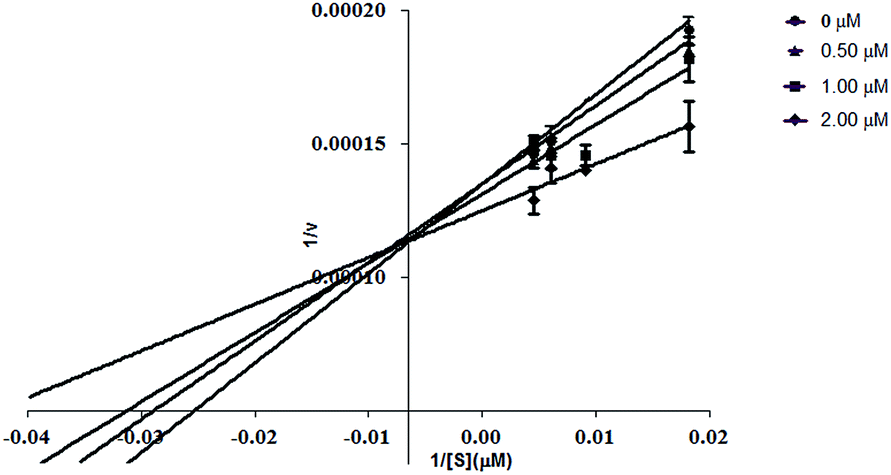 | ||
| Fig. 3 Double-reciprocal plots of the inhibition kinetics of human tissue specific intestine alkaline phosphatase (IAP) indicating competitive inhibition by compound 3e. Changes in the initial velocities of the reaction were measured at different concentrations of the inhibitor 3e using substrate CDP-Star (disodium 2-chloro-5-(4-methoxyspiro[1,2-dioxetane-3,2′-(5-chloro)tricyclo[3.3.1.13.7]decan]-4-yl)-1-phenyl phosphate). | ||
 | ||
| Fig. 4 Double-reciprocal plots of the inhibition kinetics of human tissue specific placental alkaline phosphatase (PLAP) indicating uncompetitive inhibition by compound 3e. Changes in the initial velocities of the reaction were measured at different concentrations of the inhibitor 3e using substrate CDP-Star (disodium 2-chloro-5-(4-methoxyspiro[1,2-dioxetane-3,2′-(5-chloro)tricyclo[3.3.1.13.7]decan]-4-yl)-1-phenyl phosphate). | ||
Alkaline phosphatases are metal containing enzymes and contain two zinc and one magnesium metal ion in its active site. Modelled protein structures were re-aligned with their respective templates using STAMP tool of VMD v9.1.4,31 the conservation of amino acid residues was observed. The amino acid Glu429 was found to be substituted with His434 in TNAP and with Ser429 in IAP and imparts specificity to each alkaline phosphatase. All amino acid residues forming the active site and peripheral site are fully conserved in both cases of TNAP and IAP. Fig. 5a and b shows the conservation of amino acids at active site of TNAP and IAP respectively.
 | ||
| Fig. 5 (a) Comparison of active site of template PLAP crystal structure (yellow, A) with TNAP model (purple, B). (b) Comparison of active site of template PLAP crystal structure (yellow, A) with target IAP model (blue, B). | ||
Ramachandran plot32 of TNAP and IAP models revealed good stereo-chemical quality. 95.2% of the TNAP model residues and 96.9% of IAP residues lied in favored (or core) region of torsion values. 3.1% of TNAP residues and 2.5% of IAP lie in allowed (or generous) regions of torsion values with no amino acid belonging to active site or peripheral site in this region (see ESI† for detailed Ramachandran plots of our models with each outlier labelled).
Previously, molecular docking studies of standard uncompetitive inhibitors in human TNAP revealed amino acids Glu108, Gly109, Tyr371 and His434 as major interacting amino acids. While in case of human PLAP the equivalent amino acids are Phe107, Gln108, Tyr367 and Glu429.19 The human IAP contains same amino acids as that of human PLAP with a substitution of Ser429 instead of Glu429.
Compound 3j was the most potent inhibitor of human TNAP. Binding mode of compound 3j was similar to that of PLAP. It was found to have interaction with amino acid His437 through a hydrogen bond. Amino acid His437 in TNAP is similar to His432 in PLAP, where it forms hydrogen bonding with phosphoryl oxy-moiety during substrate hydrolysis. Due to lack of positively charged group, interaction of compound 3j with Glu108 was not observed. Amino acid Arg167 forms interaction with chloride atom of the compound. Arg167 is the same amino acid as Arg166 in PLAP. Additionally, Arg119 was observed to form hydrogen bonding interaction with carboxyl moiety of our compound. Interaction of compound 3j with TNAP can be seen in Fig. 6.
 | ||
| Fig. 6 Putative binding mode of compound 3j (colored green) inside TNAP model (colored brown) and compound 3e (colored magenta) inside the crystal structure of PLAP (colored green) and inside the IAP model (colored cyan). Hydrogen bonding are shown in green colored dashed lines. | ||
Compound 3e was identified to have different modes of interaction with PLAP and IAP respectively. Due to the lack of positively charged group in compound 3e, interaction with Glu429 was not observed. It was found that compound 3e interacts with residues His432 and Gln108. Interaction of compound 3e with His432 may prevents the binding of His432 with phosphoryl oxy-moiety formed during substrate hydrolysis and cause uncompetitive inhibition of PLAP. Fig. 6 shows the interaction of compound 3e inside human PLAP enzyme.
Compound 3e in human IAP was the most potent inhibitor and the enzyme kinetic studies revealed competitive mode of inhibition unlike those found for TNAP or PLAP. The possible explanation of the competitive inhibition can be deduced from the fact that compound 3e forms a hydrogen bonding interaction with catalytic amino acid Ser92 and also binds zinc metal ion. Additionally, compound 3e was also found to have a hydrogen bonding contact with active site amino acid Arg166. As mentioned previously, alkaline phosphatase are class of enzymes belonging to serine hydrolases and thus, amino acid Ser92 plays an important role in hydrolysis of phosphomonoesters through catalytic activation by zinc ion,34 thus interaction of compound 3e with zinc and amino acid Ser92 decreases its interaction with substrate and therefore, causes competitive inhibition. Interaction of compound 3e inside the active pocket of human IAP can be seen in Fig. 6.
| Compound | Receptor | FlexX score of the top ranking pose | Binding free energy ΔG (kJ mol−1) | Poser rank |
|---|---|---|---|---|
| 3j | TNAP | −26.27 | −21 | 26 |
| 3e | PLAP | −20.19 | −12 | 08 |
| 3e | IAP | −24.50 | −12 | 07 |
Conclusions
In summary, we have explored a new class of tissue-nonspecific and tissue-specific AP inhibitors based on the quinoline template. Almost, all members of the compound library exhibited strong and effective inhibitory activity. Among them, 3j was identified as the most potent inhibitor against TNAP with an IC50 value of 0.022 ± 0.001 μM, whereas, 3e emerged as a lead candidate against IAP and PLAP activities with an IC50 value of 0.03 ± 0.01 and 0.08 ± 0.01 μM, respectively. Against GCAP, 3a was a ∼2000-fold stronger inhibitor than L-phenylalanine with an IC50 value of 0.15 ± 0.07 μM. Structure–activity relationship studies demonstrated that the presence of electron-donating and electron-withdrawing substituents as well as their substitution position imparts a marked effect on the inhibition profile. The docking study results for the tested compounds were explanatory of the enzyme inhibitory assay results, where the observed binding interactions poses clarified the binding modes and binding energy scores of these derivatives. Finally, based on all the above-mentioned investigations, this novel class of alkaline phosphatase inhibitors described here provides an opportunity for further medicinal chemistry efforts to aid in the design of second generation compounds.Experimental
Materials and methods
Unless otherwise noted, all materials were obtained from commercial suppliers (Aldrich and Merck companies) and used without further purification. Thin layer chromatography (TLC) was performed on Merck DF-Alufolien 60F254 0.2 mm precoated plates. Product spots were visualized under UV light at 254 and 365 nm. Melting points were recorded on a Stuart melting point apparatus (SMP3) and are uncorrected. Infra-red (IR) spectra were recorded on FTS 3000 MX, Bio-Rad Merlin (Excalibur model) spectrophotometer. 1H NMR spectra were recorded on a Bruker Avance (300 MHz) spectrometer. Chemical shifts (δ) are quoted in parts per million (ppm) downfield of tetramethylsilane, using residual solvent as internal standard (DMSO-d6 at 2.50 ppm). Abbreviations used in the description of resonances are: s (singlet), d (doublet), dd (doublet of doublet), t (triplet), q (quartet), m (multiplet), Ar (aromatic). Proton-decoupled 13C NMR spectra were recorded on a Bruker Avance (75 MHz) spectrometer using deuterated solvent as internal standard (DMSO-d6 at 39.52 ppm). The elemental analysis was performed on Leco CHNS-932 Elemental Analyzer, Leco Corporation (USA).Synthesis
O), 1584 (CN), 1539, 1494 (CC); 1H NMR (300 MHz, DMSO-d6): δ 14.35 (s, 1H, COOH), 8.87 (d, 1H, J = 2.4 Hz, Ar–H), 8.22 (s, 1H, Ar–H), 8.15 (d, 1H, J = 9.0 Hz, Ar–H), 7.87 (dd, 1H, J = 2.4, 2.4 Hz, Ar–H), 7.79 (dd, 1H, J = 1.2, 0.9 Hz, Ar–H), 7.67 (dd, 1H, J = 1.8, 1.8 Hz, Ar–H), 7.57–7.52 (m, 1H, Ar–H), 7.47–7.42 (m, 1H, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ 167.26, 158.76, 147.20, 140.38, 135.16, 133.57, 132.26, 132.16, 131.47, 131.16, 128.52, 125.00, 124.89, 124.71, 121.47. Analysis calcd for C16H9BrClNO2 (360.95): C, 53.00; H, 2.50; N, 3.86. Found: C, 52.88; H, 2.64; N, 3.67.O), 1589 (CN), 1547, 1499 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.73 (d, 1H, J = 2.1 Hz, Ar–H), 8.44 (s, 1H, Ar–H), 8.39 (d, 1H, J = 1.8 Hz, Ar–H), 8.19 (d, 1H, J = 7.8 Hz, Ar–H), 8.10 (d, 1H, J = 9.0 Hz, Ar–H), 7.79 (dd, 1H, J = 2.4, 2.4 Hz, Ar–H), 7.70–7.67 (m, 1H, Ar–H), 7.48 (t, 1H, J = 7.8 Hz, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ 167.68, 154.96, 147.16, 140.25, 138.21, 133.18, 132.96, 132.21, 131.51, 131.06, 130.10, 126.64, 125.04, 125.00, 122.96, 120.56. Analysis calcd for C16H9BrClNO2 (360.95): C, 53.00; H, 2.50; N, 3.86. Found: C, 52.81; H, 2.37; N, 3.72.O), 1584 (CN), 1541, 1497 (CC); 1H NMR (300 MHz, DMSO-d6): δ 14.18 (s, 1H, COOH), 8.72 (d, 1H, J = 2.3 Hz, Ar–H), 8.42 (s, 1H, Ar–H), 8.39 (d, 1H, J = 1.8 Hz, Ar–H), 8.21 (d, 1H, J = 7.8 Hz, Ar–H), 8.12 (d, 1H, J = 9.0 Hz, Ar–H), 7.70 (dd, 1H, J = 2.4, 2.4 Hz, Ar–H), 7.69–7.66 (m, 1H, Ar–H), 7.45 (t, 1H, J = 7.8 Hz, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ 175.34, 168.12, 155.13, 152.78, 150.56, 147.91, 147.79, 146.98, 135.66, 132.08, 132.00, 131.36, 130.44, 129.95, 126.92, 126.02, 124.07, 114.46, 114.20. Analysis calcd for C16H9ClFNO2 (301.03): C, 63.70; H, 3.01; N, 4.64. Found: C, 63.54; H, 2.97; N, 4.52.O), 1612 (CN), 1544, 1510 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.90 (d, 1H, J = 2.4 Hz, Ar–H), 8.19 (s, 1H, Ar–H), 8.16 (bs, 2H, Ar–H), 7.92 (d, 1H, J = 9.0 Hz, Ar–H), 7.64 (dd, 1H, J = 2.4, 2.4 Hz, Ar–H), 7.12 (d, 2H, J = 8.4 Hz, Ar–H), 2.53 (s, 3H, CH3); 13C NMR (75 MHz, DMSO-d6): δ 174.69, 168.11, 160.23, 156.38, 147.62, 147.20, 131.56, 131.38, 130.01, 128.97, 128.54, 126.90, 125.53, 121.72, 117.69, 114.21, 28.70. Analysis calcd for C17H12ClNO2: C, 68.58; H, 4.06; N, 4.70. Found: C, 68.49; H, 3.91; N, 4.61.O), 1613 (CN), 1557, 1538 (CC); 1H NMR (300 MHz, DMSO-d6): δ 8.98 (d, 1H, J = 1.8 Hz, Ar–H), 8.26 (s, 1H, Ar–H), 8.12 (d, 2H, J = 8.4 Hz, Ar–H), 7.98 (d, 1H, J = 9.0 Hz, Ar–H), 7.67–7.63 (m, 3H, Ar–H); 13C NMR (75 MHz, DMSO-d6): δ 155.35, 147.12, 138.07, 132.17, 131.49, 130.88, 130.04, 129.47, 126.90, 126.21, 123.72, 118.43. Analysis calcd for C16H9ClINO2 (408.94): C, 46.92; H, 2.21; N, 3.42. Found: C, 46.81; H, 2.12; N, 3.34.O), 1616 (CN), 1542, 1517 (CC); 1H NMR (300 MHz, DMSO-d6): δ 14.14 (s, 1H, COOH), 8.74 (d, 1H, J = 2.1 Hz, Ar–H), 8.50 (s, 1H, Ar–H), 8.18–8.10 (m, 3H, Ar–H), 7.86–7.83 (m, 1H, Ar–H), 7.36–7.31 (m, 1H, Ar–H), 3.94 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 175.29, 169.32, 155.14, 153.78, 150.56, 148.93, 148.79, 148.57, 147.08, 132.08, 132.00, 131.36, 130.44, 129.93, 126.90, 126.00, 124.07, 117.80, 114.72, 114.47, 114.20, 56.51. Analysis calcd for C17H11ClFNO3 (331.04): C, 61.55; H, 3.34; N, 4.22. Found: C, 61.32; H, 3.39; N, 4.09.O), 1583 (CN), 1557, 1538 (CC); 1H NMR (300 MHz, DMSO-d6): δ 14.18 (s, 1H, COOH), 8.95 (d, 1H, J = 2.1 Hz, Ar–H), 8.65 (d, 1H, J = 1.5 Hz), 8.17 (s, 2H, Ar–H), 7.99 (d, 1H, J = 9.0 Hz, Ar–H), 7.66 (dd, 1H, J = 2.1, 2.1 Hz, Ar–H), 7.11 (d, 1H, J = 8.7 Hz, Ar–H), 3.90 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 167.56, 153.91, 147.23, 140.47, 138.65, 133.49, 132.21, 132.11, 131.51, 131.08, 130.04, 126.84, 125.62, 125.01, 122.76, 120.56, 56.45. Analysis calcd for C17H11ClINO3 (438.95): C, 46.44; H, 2.52; N, 3.19. Found: C, 46.31; H, 2.65; N, 3.27.O), 1619 (CN), 1583, 1542 (CC); 1H NMR (300 MHz, DMSO-d6): δ 14.50 (s, 1H, COOH), 9.65 (s, 1H, OH), 8.70 (d, 1H, J = 2.4 Hz, Ar–H), 8.44 (s, 1H, Ar–H), 8.04–7.99 (m, 2H, Ar–H), 7.82–7.74 (m, 1H, Ar–H), 6.55–6.44 (m, 2H, Ar–H), 3.80 (s, 3H, OCH3), 3.75 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 167.33, 156.87, 149.60, 147.54, 140.76, 136.21, 132.66, 132.09, 131.91, 131.76, 129.10, 125.34, 125.13, 122.94, 119.43, 111.34, 56.38, 54.65. Analysis calcd for C17H12ClNO4 (343.06): C, 62.89; H, 4.10; N, 4.07. Found: C, 62.78; H, 3.98; N, 4.11.O), 1613 (CN), 1585, 1522 (CC); 1H NMR (300 MHz, DMSO-d6): δ 9.60 (s, 1H, OH), 8.71 (d, 1H, J = 2.4 Hz, Ar–H), 8.45 (s, 1H, Ar–H), 8.10 (d, 1H, J = 9.0 Hz, Ar–H), 7.87 (d, 1H, J = 1.8 Hz, Ar–H), 7.81–7.72 (m, 2H, Ar–H), 6.95 (d, 1H, J = 8.4 Hz, Ar–H), 3.92 (s, 3H, OCH3); 13C NMR (75 MHz, DMSO-d6): δ 167.81, 156.73, 149.62, 148.56, 147.34, 131.98, 130.85, 129.31, 124.84, 124.34, 121.22, 120.42, 116.25, 111.20, 56.19. Analysis calcd for C17H12ClNO4 (329.05): C, 61.92; H, 3.67; N, 4.25. Found: C, 61.99; H, 3.53; N, 4.14.Biological protocol
Homology model of the human IAP was also carried out using the same methodology as described above. The target sequence of human IAP was searched in NCBI protein database, Uniprot ID P09923 was identified. Using BLAST protein utility crystal structures of human PLAP were identified as the top ranking templates. Human placental alkaline phosphatase PDB ID 1EW2 was used, models generated were subjected to same set of evaluations as used above for human TNAP model.
Binding mode analysis
Chemical structures of compounds 3e and 3j were drawn using ACD/ChemSketch42 and 3D optimized. Using ANTECHAMBER43 utility of Chimera v.1.10. Gasteiger charges were added and the structures were energy minimized through 100 steepest descent and conjugate steps, with each step size of 0.02 Å. Compound structures were then saved as mol2 file format.
Using FlexX utility of LeadIT, docking of compounds was performed. Default docking parameters were selected and top 50 highest scoring docked positions were kept and analyzed further. Visualization of poses was performed using Discovery studio visualizer v4.0.45
Using HYDE visual affinity46 program of LeadIT the binding affinity of the docked poses were evaluated. The HYDE scoring is a function of two parameters i.e. hydrogen bond affinity and dehydration energy. Binding free energy i.e. ΔG was determined for each pose and noted down. Poses with lowest ΔG values were considered as the most stable pose with highest affinity for interaction with our receptor.
Acknowledgements
This work was supported by Higher Education Commission, Pakistan, Project No. 20-3733 and German-Pakistani Research Collaboration Programme, and by grants from the Canadian Institutes of Health Research (CIHR) to J. Sévigny. J. Sévigny was also a recipient of a “Chercheur National” research award from the Fonds de recherche du Québec – Santé (FRQS). J. Iqbal is thankful to the Organization for the Prohibition of Chemical Weapons (OPCW), The Hague, The Netherlands and Higher Education Commission of Pakistan for the financial support through Project No. 20-3733/NRPU/R&D/14/520.References
- R. B. McComb, G. N. Bowers and S. Posen, Alkaline phosphatase, Plenum Press, New York, 1979 Search PubMed.
- R. A. Stinson, J. L. McPhee and H. B. Collier, Biochim. Biophys. Acta, 1987, 913, 272–278 CrossRef CAS.
- J. R. Chan and R. A. Stinson, J. Biol. Chem., 1986, 261, 7635–7639 CAS.
- M. Lanier, E. Sergienko, A. M. Simao, Y. Su, T. Chung, J. L. Millán and J. R. Cashman, Bioorg. Med. Chem., 2009, 18, 573–579 CrossRef PubMed.
- E. A. Sergienko and J. L. Millán, Nat. Protoc., 2010, 5, 1431–1439 CrossRef CAS PubMed.
- H. van Belle, Clin. Chem., 1976, 22, 972–976 CAS.
- R. A. Mulivor, L. I. Plotkin and H. Harris, Ann. Hum. Genet., 1978, 42, 1–13 CrossRef CAS PubMed.
- J. L. Millán, Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
- S. Sidique, R. Ardecky, Y. Su, S. Narisawa, B. Brown, J. L. Millán, E. Sergienko and N. D. P. Cosford, Bioorg. Med. Chem. Lett., 2009, 19, 222–225 CrossRef CAS PubMed.
- (a) S. Narisawa, D. Harmey, M. C. Yadav, W. C. O'Neill, M. F. Hoylaerts and J. L. Millán, J. Bone Miner. Res., 2007, 22(11), 1700–1710 CrossRef CAS PubMed; (b) R. Dahl, E. A. Sergienko, Y. Su, Y. S. Mostofi, L. Yang, A. M. Simão, S. Narisawa, B. Brown, A. Mangravita-Novo, M. Vicchiarelli, L. H. Smith, W. C. O'Neill, J. L. Millán and N. D. Cosford, J. Med. Chem., 2009, 52(21), 6919–6925 CrossRef CAS PubMed; (c) L. Li, L. Chang, S. Pellet-Rostaing, F. Liger, M. Lemaire, R. Buchet and Y. Wu, Bioorg. Med. Chem., 2009, 17, 7290–7300 CrossRef CAS PubMed; (d) C. R. Sheen, P. Kuss, S. Narisawa, M. C. Yadav, J. Nigro, W. Wang, T. N. Chlea, E. Sergienko, K. Kapoor, M. R. Jackson, M. F. Hoylaerts, A. B. Pinkerton, W. C. O'Neill and J. L. Millán, J. Bone Miner. Res., 2015, 30, 824–836 CrossRef CAS PubMed.
- L. Fishman, H. Miyayama, S. G. Driscoll and W. H. Fishman, Cancer Res., 1976, 36, 2268–2273 CAS.
- W. H. Fishman, N. K. Ghosh, N. R. Inglis and S. Green, Enzymologia, 1968, 34, 317–321 CAS.
- W. H. Fishman, N. R. Inglis, S. Green, C. L. Anstiss, N. K. Gosh, A. E. Reif, R. Rustigian, M. J. Krant and L. L. Stolbach, Nature, 1968, 219, 697–699 CrossRef CAS PubMed.
- C. M. Povinelli and B. J. Knoll, Placenta, 1991, 12, 663–668 CrossRef CAS PubMed.
- B. Wahren, P. A. Holmgren and T. Stigbrand, Int. J. Cancer, 1979, 24, 749–753 CrossRef CAS.
- B. Wahren, J. Hinkula, T. Stigbrand, A. Jeppsson, L. Andersson, P. L. Esposti, F. Edsmyr and J. L. Millán, Int. J. Cancer, 1986, 37, 595–600 CrossRef CAS.
- K. A. Smans, M. B. Ingvarsson, P. Lindgren, S. Canevari, H. Walt, T. Stigbrand, T. Bäckström and J. L. Millán, Int. J. Cancer, 1999, 83, 270–277 CrossRef CAS.
- C. E. Ovitt, A. W. Strauss, D. H. Alpers, J. Y. Chou and I. Boime, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 3781–3785 CrossRef CAS.
- A. Kozlenkov, M. F. Hoylaerts, T. Ny, M.-H. Le Du and J. L. Millán, J. Bone Miner. Res., 2004, 19, 1862–1872 CrossRef CAS PubMed.
- R. J. Ardecky, E. V. Bobkova, T. Kiffer-Moreira, B. Brown, S. Ganji, J. Zou, I. Pass, S. Narisawa, F. G. Iano, C. Rosenstein, A. Cheltsov, J. Rascon, M. Hedrick, C. Gaslor, A. Forster, S. Shi, R. Dahl, S. Vasile, Y. Su, E. Sergienko, T. C. Chung, J. Kaunitz, M. F. Hoylaerts, A. B. Pinkerton and J. L. Millán, Bioorg. Med. Chem. Lett., 2014, 24(3), 1000–10004 CrossRef CAS PubMed.
- S. Andrews, S. J. Burgess, D. Skaalrud, J. X. Kelly and D. H. Peyton, J. Med. Chem., 2010, 53, 916–919 CrossRef CAS PubMed.
- (a) R. S. Upadhayaya, J. K. Vandavasi, R. A. Kardile, S. V. Lahore, S. S. Dixit, H. S. Deokar, P. D. Shinde, M. P. Sarmah and J. Chattopadhyaya, Eur. J. Med. Chem., 2010, 45, 1854–1867 CrossRef CAS PubMed; (b) S. Eswaran, A. V. Adhikari and R. A. Kumar, Eur. J. Med. Chem., 2010, 45, 957–966 CrossRef CAS PubMed; (c) C. L. Yang, C. H. Tseng, Y. L. Chen, C. M. Lu, C. L. Kao, H. Y. Tseng, M. H. Wu and C. C. Tzeng, Eur. J. Med. Chem., 2010, 45, 602–607 CrossRef CAS PubMed.
- (a) C. H. Tseng, C. C. Tzeng, C. L. Yang, P. J. Lu, H. L. Chen, H. Y. Li, Y. C. Chuang, C. N. Yang and Y. L. Chen, J. Med. Chem., 2010, 53, 6164–6179 CrossRef CAS PubMed; (b) C. H. Tseng, Y. L. Chen, C. L. Yang, C. M. Cheng, C. H. Han and C. C. Tzeng, Bioorg. Med. Chem., 2012, 20, 4397–4404 CrossRef CAS PubMed; (c) C. H. Tseng, Y. L. Chen, C. Y. Hsu, T. C. Chen, C. M. Cheng, H. C. Tso, Y. J. Lu and C. C. Tzeng, Eur. J. Med. Chem., 2013, 59, 274–282 CrossRef CAS PubMed.
- K. Kaur, M. Jain, R. P. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS PubMed.
- (a) N. Ahmed, K. G. Brahmbhatt, S. Sabde, D. Mitra, I. P. Singh and K. K. Bhutani, Bioorg. Med. Chem., 2010, 18, 2872–2879 CrossRef CAS PubMed; (b) H. K. Peng, C. K. Lin, S. Y. Yang, C. K. Tseng, C. C. Tzeng, J. C. Lee and S. C. Yang, Bioorg. Med. Chem. Lett., 2012, 22, 1107–1110 CrossRef CAS PubMed.
- (a) P. Labrie, S. P. Maddaford, J. Lacroix, C. Catalano, D. K. Lee, S. Rakhit and R. C. Gaudreault, Bioorg. Med. Chem., 2007, 15, 3854–3868 CAS; (b) M. Naito, Y. Matsuba, S. Sato, H. Hirata and T. Tsuruo, Clin. Cancer Res., 2002, 8, 582–588 Search PubMed.
- X. Ji, H. Huang, Y. Li, H. Chen and H. Jiang, Angew. Chem., Int. Ed., 2012, 51, 7292–7296 CrossRef CAS PubMed.
- (a) M. al-Rashida, R. Raza, G. Abbas, M. S. Shah, G. E. Kostakis, J. Lecka, J. Sévigny, M. Muddassar, C. Papatriantafyllopoulou and J. Iqbal, Eur. J. Med. Chem., 2013, 66, 438–449 CrossRef CAS PubMed; (b) M. al-Rashida, S. A. Ejaz, S. Ali, A. Shaukat, M. Hamayoun, M. Ahmed and J. Iqbal, Bioorg. Med. Chem., 2015, 23(10), 2435–2444 CrossRef CAS PubMed.
- P. Das, X. Deng, L. Zhang, M. G. Roth, B. M. A. Fontoura, M. A. Phillips and J. K. de Brabander, ACS Med. Chem. Lett., 2013, 4, 517–521 CrossRef CAS PubMed.
- S. Narisawa, D. Harmey, M. C. Yadav, W. C. O'Neill, M. F. Hoylaerts and J. L. Millán, J. Bone Miner. Res., 2007, 22, 1700–1710 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- S. C. Lovell, I. W. Davis, W. B. Arendall III, P. I. W. de Bakker, J. M. Word, M. G. Prisant, J. S. Richardson and D. C. Richardson, Proteins: Struct., Funct., Genet., 2003, 50, 437–450 CrossRef CAS PubMed.
- K. Chen, K. Wang, A. M. Kirichian, A. F. Al Aowad, L. K. Iyer, S. J. Adelstei and A. I. Kassis, Mol. Cancer Ther., 2006, 5(12), 3001–3013 CrossRef CAS PubMed.
- P. Llinas, E. A. Stura, A. Ménez, Z. Kiss, T. Stigbrand, J. L. Millán and M. H. le Du, J. Mol. Biol., 2005, 350(3), 441–451 CrossRef CAS PubMed.
- F. Kukulski, S. A. Lévesque, E. G. Lavoie, J. Lecka, F. Bigonnesse, A. F. Knowles, S. C. Robson, T. L. Kirley and J. Sévigny, Purinergic Signalling, 2005, 1(2), 193–204 CrossRef CAS PubMed.
- T. Kiffer-Moreira, C. R. Sheen, K. C. da Silva Gasque, M. Bolean, P. Ciancaglini, A. van Elsas, M. F. Hoylaerts and J. L. Millán, PLoS One, 2014, 9(2), e89374 Search PubMed.
- R. J. Ardecky, E. V. Bobkova, T. Kiffer-Moreira, B. Brown, S. Ganji, J. W. Zou, I. Pass, S. Narisawa, F. G. Iano, C. Rosenstein, A. Cheltsov, J. Rascon, M. Hedrick, C. Gasior, A. Forster, S. H. Shi, R. Dahl, S. Vasile, Y. Su, E. Sergienko, T. D. Y. Chung, J. Kaunitz, M. F. Hoylaerts, A. B. Pinkerton and J. L. Millán, Bioorg. Med. Chem. Lett., 2014, 24(3), 1000–1004 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234(3), 779–815 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrine, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215(3), 403–410 CrossRef CAS PubMed.
- ACD/Chemsketch, 2014, at http://www.acdlabs.com.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25(2), 247–260 CrossRef CAS PubMed.
- LeadIT, 2014, at http://www.biosolveit.de/LeadIT/.
- Accelrys Software Inc., Discovery Studio Modeling Environment, Release 4.0, San Diego, Accelrys Software Inc., 2013 Search PubMed.
- N. Schneider, G. Lange, S. Hindle, R. Klein and M. Rarey, J. Comput.-Aided Mol. Des., 2013, 27(1), 15–29 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12455g |
| ‡ These authors contributed equally. |
| § Present address: School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD (UK). |
| This journal is © The Royal Society of Chemistry 2015 |