
Hierarchically porous polystyrene membranes fabricated via a CO2-expanded liquid selective swelling and in situ hyper-cross-linking method†
Abstract
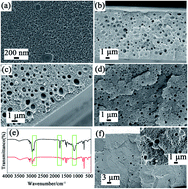
Hierarchically porous polymeric materials represent a new class of materials that have attracted both industrial and academic interest. This paper presents a novel, etching-free and versatile preparation methodology, using commercially available polystyrene and a CO2-expanded liquid selective swelling process combined with a hyper-cross-linking reaction. The morphology of the membranes was observed with electron microscopes, and the chemical structure of the membranes was investigated using Fourier transform infrared spectrometry and solid-state nuclear magnetic resonance measurements. One level of macroporous structures was produced by a CO2-expanded methanol selective swelling process, while the other level of micropores was created via the hyper-cross-linking reaction. The cross-linked membranes possessed large specific surface areas and excellent thermal stability, and have potential applications in catalysis, separation and gas storage.
 Please wait while we load your content...
Please wait while we load your content...

