
Low temperature synthesis of ZrS2 nanoflakes and their catalytic activity†
Yan Wen,
Yanqiu Zhu and
Shaowei Zhang*
College of Engineering, Mathematics and Physical Sciences, University of Exeter, Exeter, EX4 4QF, UK. E-mail: s.zhang@exeter.ac.uk
First published on 27th July 2015
Abstract
ZrS2 nanoflakes with a narrow size distribution (10–30 nm) were successfully prepared via a direct gaseous reaction between ZrCl4 and S at a relatively low temperature (800 °C). They exhibited unique optical properties and showed excellent performance in photocatalytic degradation of 4-nitrophenol, one of the most-difficult-to-remove pollutants.
During the past two decades, transition metal chalcogenides have attracted a great deal of attention because of their excellent optical, electrical, mechanical and catalytic properties.1–5 They can be used or potentially used in many important areas including in batteries, lubricants, drug carriers, and dye-sensitised solar cells (DSCs).6–9 Among transition metal chalcogenides, ZrS2 is a semiconductor with a graphite-like layered structure. Because of this structure, it is possible to prepare various nanostructured ZrS2 materials such as nanoflakes, nanobelts and nanotubes.10,11 These nanostructured ZrS2 materials exhibit unique physical and chemical properties originating from a quantum-confinement effect and thus are regarded as good candidate materials for making various novel nanodevices, e.g., field emitters.11
To synthesise these nanostructured ZrS2, a few techniques have been attempted, of which the most straightforward one was to directly heat Zr and S powders at 850 °C in an inert atmosphere. In this case, S vapour generated from the original solid S reacted with solid Zr, forming ZrS2 nanobelts possessing good electrical properties.12 By replacing S with CS2, the reaction temperature could be further reduced to 800 °C.12 Another synthesis technique was via decomposition of ZrS3 at 900 °C in H2.13 ZrS2 nanotubes with about 140 nm in diameter were resulted.
Recently, a few types of monolayered or multi-layered transition metal chalcogenides have been prepared. They exhibited enhanced electrical and mechanical properties compared to their bulk counterparts.14 By reacting ZrCl4, oleylamine and CS2, about 1.6 nm thick ZrS2 nanodiscs with controllable diameters (20–60 nm) were successfully produced. These nanodiscs exhibited good performance as an anodic material for Li+ intercalation in Li-ion batteries. Compared to the case using bulk ZrS2, the discharge capacity was enhanced by 230%, along with much improved stability.15 Nevertheless, this technique still suffers from several drawbacks, including the usage of hazard raw materials (in particular CS2) and complicated process.
To overcome the drawbacks of the synthesis techniques reported to date, a new synthesis technique was developed in this work. ZrS2 nanoflakes were prepared via a gaseous reaction process between ZrCl4 and S at a relatively low temperature (800 °C). Furthermore, photocatalytic properties of as-prepared ZrS2 nanoflakes were explored for the first time. The results indicated that as-prepared ZrS2 nanoflakes exhibited excellent catalytic activity in photocatalytic decomposition of 4-nitrophenol (4-NP), one of the most-difficult-to-remove pollutants inevitably present in pesticides, herbicides, insecticides, synthetic dyes, etc.
Preparation of ZrS2 nanoflakes
ZrCl4 (>99.5% Sigma-Aldrich) and S (99% Sigma-Aldrich) powders were used as the starting raw materials. A quartz tube in an electric furnace was flushed with Ar at 40 mL min−1 for 30 min to remove any residual air and was then heated at 10 °C min−1 to a given temperature between 650–800 °C. During heating, H2 was introduced into the tube at 20 mL min−1 along with Ar flowing at 40 mL min−1. A mixture of S and ZrCl4 in the molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was placed in a ceramic boat and pushed quickly into the hot zone of the furnace preheated to the given temperature and held for 1 h. After furnace cooling to room temperature, the reacted mass was collected from the ceramic boat and subjected to further characterisation.
:1 was placed in a ceramic boat and pushed quickly into the hot zone of the furnace preheated to the given temperature and held for 1 h. After furnace cooling to room temperature, the reacted mass was collected from the ceramic boat and subjected to further characterisation.
Characterisation
Phase analysis on the reacted powders was performed using an X-ray diffractometer (D500 Siemens) at 30 mA and 40 kV with Ni-filtered Cu Kα radiation (λ = 0.154 nm). The scan rate (2θ) of 2° min−1 with a step size of 0.02° was used within the scan range of 10–80°. A JEOL 2100 TEM at an accelerating voltage of 200 kV was used to observe microstructures of the reacted samples. UV-vis spectra and photoluminescence excitation spectra were recorded by a Jenway 6715 UV/visible spectrophotometer and a Hitachi F-4500 fluorescence spectrophotometer respectively. A small amount of sample was dispersed in water and examined at room temperature.Catalytic decomposition of 4-NP
As-prepared ZrS2 nanoflakes were ultrasonically dispersed in 10 mL distilled water for 30 min to form a homogenous solution/suspension of 0.5 × 10−3 mol L−1 which was further combined with 10 mL 3.2 × 10−3 mol L−1 4-NP aqueous solution and 1 mL H2O2 solution (30% in weight) under stirring. The resultant solution/suspension was exposed to the beam generated from a Xenon lamp at room temperature. The reaction progress was monitored by UV-vis measurements on 1 mL solution taken out every 30 min from the reacting solution.| ZrCl4 + 3S + 2H2 = ZrS3 + 4HCl | (1) |
| ZrS3 + H2 = ZrS2 + H2S | (2) |
Fig. 1 presents XRD patterns of samples resultant from 1 h firing at 650, 700, 750 and 800 °C. At 650 °C, ZrS3 rather than ZrS2 was formed as the primary phase (Fig. 1a), indicating occurrence of the reaction between ZrCl4, S and H2 (Reaction (1)). As excessive S was used to compensate for its high temperature evaporation, Reaction (1) was essentially proceeded in an S-rich atmosphere. In such an S-rich atmosphere and at this relatively low temperature, ZrS3 is thermodynamically stable.11 Upon increasing the temperature to 700 °C, small amounts of ZrS2 were detected along with the primary phase of ZrS3 (Fig. 1b), indicating that ZrS3 was not stable at this temperature and started to decompose into ZrS2 according to Reaction (2). Upon further increasing the temperature to 750 °C, ZrS2 became the primary phase, and only minor ZrS3 remained (Fig. 1c). Finally, when the temperature increased to 800 °C, only ZrS2 was formed and no other phases were detected (Fig. 1d).
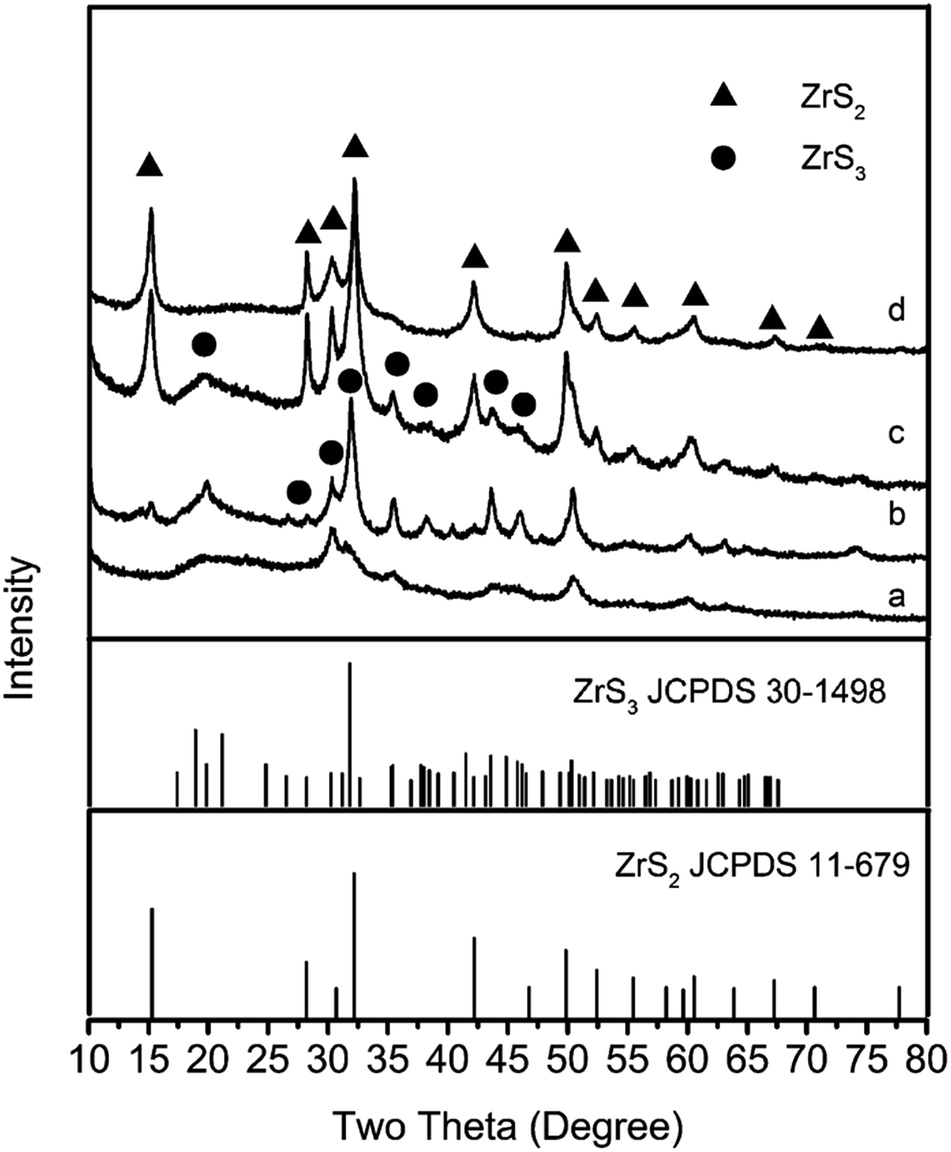 | ||
| Fig. 1 XRD patterns of samples resulting from 1 h firing in H2/Ar at (a) 650 °C, (b) 700 °C, (c) 750 °C and (d) 800 °C. | ||
Fig. 2 presents TEM images of some of the samples whose XRD patterns are shown in Fig. 1. The ZrS3 nanosheets resultant from 1 h firing at 650 °C (Fig. 2a) were nearly transparent, implying their very small thicknesses. The average size and thickness of the curved nanosheets were measured to be approximately 150 and 7 nm, respectively. Due to their small thicknesses and relatively high surface areas, the ZrS3 nanosheets tended to curve and connect with each other to maintain the minimum thermodynamic energy,16 which explained the agglomerations among them. At 700 °C, the formed ZrS3 nanosheets began to disintegrate into small nanoflakes with sizes below 100 nm (Fig. S1†). Upon increasing the temperature to 750 °C, these generated nanoflakes were further reduced by H2 (Reaction (2)). The loss of S during the reduction caused crushing of the initial nanosheets into tiny fragments,11 which finally crystallised to ZrS2 (Fig. 2b). ZrS2 particles synthesised at 800 °C had angular shapes and a narrow size distribution (10–30 nm) (Fig. 2c). An HRTEM image (Fig. 2d) further reveals that the ZrS2 nanoflakes were well crystallised, with a lattice spacing of 0.31 nm which matches with the spacing between the (100) planes of ZrS2 (Fig. 2d).
 | ||
| Fig. 2 TEM images of samples resultant from 1 h firing at (a) 650 °C, (b) 750 °C and (c & d) 800 °C in Ar/H2 atmosphere. | ||
Based on the results shown in Fig. 1 and the discussion above, the overall reaction process could be summarised as follows: ZrS3 nanosheets were initially formed from the reaction between ZrCl4 and S and remained stable until 650 °C (Fig. 1a). At >650 °C, ZrS3 nanosheets would decompose to form ZrS2 nanoflakes (Fig. 1b and c and 2a), and with increasing the temperature, the decomposition rate increased. Upon the completion of the reaction at 800 °C, all of the ZrS3 had been consumed and phase pure ZrS2 nanoflakes were obtained (Fig. 1d and 2c and d).
Presented in Fig. 3 are the UV-vis, PL and PLE spectra of the ZrS2 nanoflakes resultant from 1 h firing at 800 °C in H2/Ar. Two peaks centred at 245 nm and 280 nm are observed (Fig. 3a). Compared with the characteristic UV-vis absorption peak of a ZrS2 thin film (centred at approximately 400 nm),17 the absorption peak in Fig. 3a exhibited obvious blue-shift, arising from the strong quantum confinement effect. According to Fig. 3b, several fluorescence spectra were measured with various excitation wavelengths between 290 and 320 nm. The strongest emissions of the ZrS2 nanoflakes occurred at 330 and 356 nm (UV area) when a 290 nm excitation wavelength was used. Upon changing the excitation wavelength, the fluorescence band position remained unchanged, indicating that the fluorescence was generated under the same initial and final states even when the excitation wavelength varied between 290 and 320 nm. This effect was attributed to the fast energy relaxation from the excitation state, which was excited by the photo-excitation to the ground state. A strong peak (∼291 nm) and a weak peak (∼229 nm) appeared in the photoluminescence-excitation (PLE) spectrum of the ZrS2 nanoflakes with a detection wavelength of 330 nm (Fig. 3c). Although ZrS2 is an indirect gap semiconductor, it could be transferred to a direct gap semiconductor upon decreasing the atomic layers.18 The evident luminescence of as-synthesised ZrS2 nanoflakes (Fig. 4b) indicated such an indirect-to-direct band structure change, which was consistent with the thin-layered structure observed by HRTEM (Fig. 2d). The two excitation peaks in the PLE spectrum could be assigned to the following: (1) excitation from the V2 point to the conduction band minimum (band gap ∼4.43 eV) and (2) excitation from the V1 point to the conduction band minimum (band gap ∼5.06 eV). These band gaps were much larger than those reported for monolayer ZrS2 (∼2 eV)18 because of the strong quantum confinement of the as-synthesised ZrS2 nanoflakes. This wide band gap could efficiently hinder the recombination of excited electrons and thus extend the lifetime of electron holes, leading to improved oxidation performance.
 | ||
| Fig. 3 (a) UV-vis spectrum of as-synthesised ZrS2 nanoflakes from 1 h firing at 800 °C. (b) PL spectrum of as-synthesised ZrS2 nanoflakes with various excitation wavelengths. (c) Corresponding PLE spectrum with 330 nm detection wavelength. (d) Diagram of the band structure of as-synthesised ZrS2 nanoflakes close to the K point (V1 and V2 are the two valence band maxima). | ||
 | ||
| Fig. 4 (a) Evolution of UV-vis absorption spectrum with time, in the case of ZrS2 catalysed degradation of 4-NP. (b) Plot of ln(Ct/C0) versus time, in the case of using ZrS2 as the catalyst. | ||
Some semiconductor materials such as TiO2 and ZnO have been reported to exhibit strong photocatalytic activities.19,20 When they are irradiated with light of a suitable wavelength, electrons will jump from the valence band (VB) to the conduction band (CB), leaving holes behind. In an aqueous suspension, the electron holes generated can react with OH− groups to form ˙OH radicals which are very strong oxidising species21,22 and thus can readily oxidise some organic compounds. Since the as-synthesised ZrS2 nanoflakes had a large band gap (4.43 eV), excited electrons need a relatively long time to refill the electron holes, extending the lifetime of ˙OH radicals, and leading to strong oxidation. In this work, H2O2 was used as an electron acceptor to enhance the oxidation process because of two main reasons. One was that H2O2 could accept excited electrons, generating more radical ˙OH radicals, and the other was that the acceptance of excited electrons by H2O2 could inhibit the recombination of electrons and holes, thus further improving the oxidation performance.23 Fig. 4 illustrates the UV-vis spectrum evolution of the 4-NP degradation with time. The 4-NP–H2O2 solution gave a UV absorption peak centred at approximately 320 nm (the characteristic peak of 4-NP20). When ZrS2 nanoflakes were used, the peak height at 320 nm evidently decreased with time. The ratio between the 4-NP concentrations at times t (Ct) and 0 (C0) was measured from the relative intensity ratio of the respective absorbance, At/A0, at 320 nm. A linear relationship between ln(Ct/C0) and time was obtained (Fig. 4b), indicating a first-order reaction. Standard deviation from the mean was determined to estimate the error in ln(Ct/C0). From the slope of the straight line in Fig. 4b, the reaction rate constant when using ZrS2 nanoflakes as a catalyst was calculated to be 2.22 × 10−3 min−1. To compare, photocatalytic degradation tests were also carried out on the 4-NP/H2O2 solutions without using ZrS2 nanoflakes. According to Fig. S2,† very little degradation was observed after 1 h, indicating that H2O2 itself did not show any obvious catalytic effects on the photocatalytic degradation of 4-NP. The above results indicated that as-synthesised ZrS2 showed excellent catalytic activity in the decomposition of 4-NP, one of the most-difficult-to-remove pollutants commonly present in many systems, thus could be potentially used as a promising catalyst for future pollutant purifications.
Conclusions
ZrS2 nanoflakes were produced via a direct gaseous reaction between ZrCl4 and S at a relatively low temperature (800 °C). The generated ZrS2 nanoflakes had a narrow size distribution (10–30 nm). During the heat treatment, thin ZrS3 nanosheets were initially formed at 650 °C but decomposed into ZrS2 nanoflakes upon increasing the temperature to above 650 °C. Because of their small sizes, as-prepared ZrS2 nanoflakes exhibited strong quantum confinement effect and a large band gap (4.43 eV). Moreover, they also exhibited excellent photocatalytic activity in the degradation of 4-NP.Notes and references
- X. F. Duan, Y. Huang, R. Agarwal and C. M. Lieber, Nature, 2003, 421, 241–245 CrossRef CAS PubMed.
- M. R. Gao, Y. F. Xu, J. Jiang and S. H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- J. Chen and F. Wu, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 989–994 CrossRef CAS.
- G. L. Frey, S. Elani, M. Homyonfer, Y. Feldman and R. Tenne, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 6666–6671 CrossRef CAS.
- O. Tevet, O. Goldbart, S. R. Cohen, R. Rosentsveig, R. Popovitz-Biro, H. D. Wagner and R. Tenne, Nanotechnology, 2010, 21, 365705 CrossRef CAS PubMed.
- Y. L. Liang, R. J. Feng, S. Q. Yang, H. Ma, J. Liang and J. Chen, Adv. Mater., 2011, 23, 640–643 CrossRef CAS PubMed.
- H. B. Yang, S. K. Liu, J. X. Lil, M. H. Li, G. Peng and G. T. Zou, Nanotechnology, 2006, 17, 1512–1519 CrossRef CAS.
- H. Wu, R. Yang, B. Song, Q. Han, J. Li, Y. Zhang, Y. Fang, R. Tenne and C. Wang, ACS Nano, 2011, 5, 1276–1281 CrossRef CAS PubMed.
- H. C. Sun, D. Qin, S. Q. Huang, X. Z. Guo, D. M. Li, Y. H. Luo and Q. B. Meng, Energy Environ. Sci., 2011, 4, 2630–2637 CAS.
- A. H. Reshak and S. Auluck, Phys. B, 2004, 353, 230–237 CrossRef CAS PubMed.
- Y. L. Zhang, X. C. Wu, Y. R. Tao, C. J. Mao and J. J. Zhu, Chem. Commun., 2008, 2683–2685 RSC.
- B. T. Kaminskii, G. N. Prokof'eva, A. S. Plygunov and P. A. Galitskii, Powder Metall. Met. Ceram., 1973, 12, 521–524 CrossRef.
- M. Nath and C. N. R. Rao, Angew. Chem., Int. Ed., 2002, 41, 3451–3454 CrossRef CAS.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- J. T. Jang, S. Jeong, J. W. Seo, M. C. Kim, E. Sim, Y. Oh, S. Nam, B. Park and J. Cheon, J. Am. Chem. Soc., 2011, 133, 7636–7639 CrossRef CAS PubMed.
- Y. D. D. Li, X. L. L. Li, R. R. R. He, J. Zhu and Z. X. X. Deng, J. Am. Chem. Soc., 2002, 124, 1411–1416 CrossRef CAS PubMed.
- M. M. B. R. Thiyagarajan and M. Anusuya, J. Am. Sci., 2009, 5, 6–12 Search PubMed.
- Y. Li, J. Kang and J. B. Li, RSC Adv., 2014, 4, 7396–7401 RSC.
- G. Mele, E. Garcia-Lopez, L. Palmisano, G. Dyrda and R. Slota, J. Phys. Chem. C, 2007, 111, 6581–6588 CAS.
- S. T. Kochuveedu, Y. H. Jang, Y. J. Jang and D. H. Kim, J. Mater. Chem. A, 2013, 1, 898–905 CAS.
- J. C. Doliveira, C. Minero, E. Pelizzetti and P. Pichat, J. Photochem. Photobiol., A, 1993, 72, 261–267 CrossRef CAS.
- R. W. Matthews and S. R. Mcevoy, J. Photochem. Photobiol., A, 1992, 64, 231–246 CrossRef CAS.
- G. Mele, G. Ciccarella, G. Vasapollo, E. Garcia-Lopez, L. Palmisano and M. Schiavello, Appl. Catal., B, 2002, 38, 309–319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12412c |
| This journal is © The Royal Society of Chemistry 2015 |