DOI:
10.1039/C5RA12407G
(Paper)
RSC Adv., 2015,
5, 77965-77972
A highly selective chemosensor for naked-eye sensing of nanomolar Cu(II) in an aqueous medium†
Received
26th June 2015
, Accepted 1st September 2015
First published on 1st September 2015
Abstract
A novel highly selective and sensitive colorimetric chemosensor L for the detection of Cu2+ ion with a fast response time was designed and synthesized. Receptor L detected Cu2+ ion by changing its color from colorless to magenta in a semi-aqueous solution. The limit of detection for Cu2+ was calculated to be as low as 28 nM. The possible binding mode of compound L with Cu2+ ion was studied using the Job's method, HRMS, FTIR spectroscopy and 1H NMR spectroscopy titration. Importantly, test strips containing L were fabricated as a naked-eye indicator for Cu2+ ion in pure water samples.
Introduction
The recognition of biologically and chemically important species at ultra-low concentrations has received considerable attention in recent decades due to their important roles in biological and environmental processes, ranging from the diagnosis of life-threatening diseases to analysis of environmental pollutants.1 Copper ion, an indispensible transition metal ion in the human body, plays various roles in physiological processes and is a key component of a wide range of enzymes such as copper–zinc superoxide dismutase, cytochrome c oxidase, ceruloplasmin, lysyl oxidase, tyrosinase, dopamine b-hydroxylase and peptidylglycine a-amidating monooxygenase.2 Aberrations in normal copper levels, both systemic as well as on a tissue or cellular scale, are implicated in a wide range of diseases such as Menkes disease, Wilson's disease, Alzheimer's disease, Parkinson's disease and transmissible spongiform encephalopathy (prion diseases)2(a),3. On the other hand, Cu2+ is a significant environmental pollutant throughout the world due to its widespread use in industry, agriculture, household utensils and water pipes. Under normal conditions, the average concentration of copper in the blood should not exceed 100–150 μg dL−1 (15.7–23.6 μM).4 Therefore, it is of increasing importance to develop fast, convenient and reliable methods for the qualitative and quantitative detection of trace amounts of copper ion in light of its biological and environmental implications.
To date, a large amount of study has been reported for the detection of Cu2+ ions at trace levels, and a series of conventional analytical methods, such as inductively coupled plasma atomic emission/mass spectroscopy (ICP-AES/ICP-MS),5 atomic absorption spectrometry (AAS),6 electrochemical methods,7 surface plasmon resonance detectors8 and quantum-dot-based assays,9 have been developed. Although these technologies can detect Cu2+ ions selectively with high sensitivity, they require highly sophisticated/expensive instrumentation and time-consuming processes, requiring tedious sample preparation and highly trained operators, which means they cannot be used for real-time detection in their routine application.10
Because of the inexpensiveness, high sensitivity and simplicity, fluorescence techniques have attracted widespread attention in the recent years, and a rapidly increasing number of metal-responsive fluorescent sensors have been studied.11 However, unfortunately, Cu2+ is a notorious fluorescence quencher because of its paramagnetic nature.12 Therefore, many of the reported Cu2+ sensors undergo fluorescence quenching upon binding of Cu2+ either by electron or energy transfer mechanisms.13 In addition, fluorescence techniques sometimes still require tedious sample preparation procedures and trained operators for bio-imaging research (i.e., preparation of buffer solution with different types and dosage, choice of cell resources and cell culture).
In contrast, unlike the abovementioned techniques, colorimetric methods14 based on color changes appeared to be the most attractive technique since they can conveniently and easily monitor target ions directly by the naked eye even at the micro/submicromolar levels without any need for expensive and/or sophisticated instrumentation. Upon surveying the literature, many relevant works concerning colorimetric Cu2+ probes have been reported,15 and we noticed that most of the reported Cu2+ selective colorimetric sensors have a number of drawbacks (i.e., poor detection limit, long response time and interference from other transition metal ions) (Table 1)15(e)–(g),16. Therefore, exploring more excellent colorimetric chemosensing molecules for the naked-eye detection of Cu2+ in an aqueous solution is still in demand. Studies related to this area are of great challenge and continue to be of widespread interest.
Table 1 Comparison of the reported colorimetric chemosensors for naked-eye detection of Cu(II)
| Chemosensors |
Detection limit (μM) |
Water content of solution |
Interference |
Response time |
References |
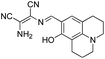 |
2.1 |
40% |
None |
No data |
15e |
 |
3.42 |
90% |
None |
Less than 30 s |
15f |
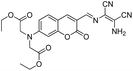 |
1.2 |
60% |
Hg2+, Fe3+ |
No data |
15g |
| Cys-modified AuNR (Cys-AuNR) |
0.34 |
100% |
Hg2+ |
5 min |
16a |
 |
2.7 |
100% |
None |
No data |
16b |
 |
No data |
20% |
No data |
No data |
16c |
 |
2.29 |
80% |
None |
No data |
16d |
 |
13.6 |
10% |
None |
No data |
16e |
 |
0.028 |
50% |
Hg2+ |
1 min |
This study |
In this study, we successfully synthesized and characterized a simple colorimetric chemosensor L, as depicted in Scheme 1. Intriguingly, L gives a visual color change from colorless to magenta with a fast response time, allowing for the naked-eye detection of Cu2+ with high selectivity and sensitivity in a DMSO–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solution. The sensing behavior of L towards Cu2+ was investigated systematically. Importantly, test strips were prepared as a practical, visible colorimetric detection kit for Cu(II). Using the remarkable colorimetric response of L to Cu(II), a recreational test was performed successfully.
:1, v/v) solution. The sensing behavior of L towards Cu2+ was investigated systematically. Importantly, test strips were prepared as a practical, visible colorimetric detection kit for Cu(II). Using the remarkable colorimetric response of L to Cu(II), a recreational test was performed successfully.
 |
| | Scheme 1 Synthetic procedure of L. | |
Experimental
Materials and measurements
All starting materials were purchased from commercial suppliers and used without further purification. Deionized water was used throughout the experiments. NMR spectra were obtained on a Varian 400 MHz NMR spectrometer using CDCl3 or DMSO-d6 as the solvent and TMS as the internal standard. 1H NMR titration experiments were carried out in DMSO-d6. A UV-1800 UV-Vis spectrophotometer with 1.0 cm quartz cell was used to record the absorbance measurements. HRMS were determined on a LCMS-IT from a Shimadzu TOF (LC30A). FTIR spectra were obtained on a Bruker TENSOR27, Germany. The purity of L–Cu2+ complex was determined by elemental analysis performed on a EuroEA Elemental Analyser. TLC analysis was performed on silica gel plates (GF254, model number; 0.20–0.25 mm, thickness) and column chromatography was conducted over a silica gel (100–200, mesh size), both of which were obtained from Qingdao Ocean Chemicals.
General methods
A stock solution of L (0.2 mM) was prepared in DMSO. Metal ion solutions (10 mM) of KCl, Co(NO3)2·6H2O, Ni(NO3)2·6H2O, Zn(NO3)2, Fe(NO3)3·9H2O, MgSO4·7H2O, Al(ClO4)3·9H2O, Pb(NO3)2, Mn(NO3)2, HgCl2, AgNO3, Ca(ClO4)2, NaCl, La(NO3)3·6H2O, Cd(NO3)2·4H2O, FeSO4·7H2O, CuCl2·2H2O, CuSO4·5H2O, Cu(NO3)2·3H2O and Cu(OAc)2·H2O and organic or inorganic anion solutions (10 mM) of (CH3CH2CH2CH2)4N+F−, (CH3CH2CH2CH2)4N+Cl−, (CH3CH2CH2CH2)4N+Br−, (CH3CH2CH2CH2)4N+I−, CH3COONa·3H2O, NaNO3, Na2CO3, NaHCO3, Na2SO4, NaHSO4·H2O, Na2HPO4·12H2O, NaH2PO4·2H2O and Na2S2O3·5H2O were prepared in deionized water. The solution of L (0.5 mL) was diluted to 10 μM with DMSO and deionized water in a 10 mL volumetric flask and then the ions were added. Spectral data were recorded after a 1 min of incubation period.
Determination of the detection limit
The detection limit was calculated with the following formula17 based on the absorbance titration:
where σ is the standard deviation of blank measurements and k is the slope between the absorption intensity at 562 nm versus Cu2+ concentration. The absorbance intensity of the blank L (10 μM) was measured 10 times in a DMSO–water (1:1, v/v) solution.
Determination of the association constant
The association constant of L–Cu2+ complex was determined from the Benesi–Hildebrand equation:18
where K denotes the association constant, A0 is the observed absorption in the absence of cation, A is the observed absorption with added cation, [M] is the concentration of the cation added and a and b are constants. The association constant value K was evaluated graphically by plotting 1/ΔA against 1/[M].
Synthesis of rhodamine B hydrazide (1)
Rhodamine B hydrazide (1) was synthesized as previously reported.19
Synthesis of 6-hydroxy-4-methylcoumarin. 6 mL of concentrated sulfuric acid was placed in a 50 mL round-bottomed flask in an ice bath followed by the dropwise addition of a solution containing hydroquinone (1.50 g, 13.6 mmol) and excess ethyl acetoacetate (4 mL, 31.6 mmol). The mixture was stirred at 5–10 °C for 12 h and then warmed to room temperature. The resultant dark yellow solution was poured into crushed ice with vigorous stirring. The precipitate was collected by suction filtration, washed several times with cold water, and dried in vacuum to obtain a canary yellow solid (0.89 g, yield: 38%).
Synthesis of 7-formyl-6-hydroxy-4-methylcoumarin (2). 6-Hydroxy-4-methylcoumarin (1.00 g, 5.68 mmol) and hexamethylenetetramine (1.98 g, 14.12 mmol) were placed in a 50 mL schlenk flask, 10 mL of trifluoroacetic acid was added under a dry argon atmosphere and the mixture was kept in an ice bath for 30 min. After warming to room temperature, the mixture was refluxed at 100 °C in an argon protection environment for 14 h. The solvent was evaporated under reduced pressure and the residual solution was poured into 100 mL of cold deionized water with stirring. The precipitate was collected by suction filtration, repeatedly washed with cold water and dried in vacuum to afford a yellow powder. The crude product was further purified through silica gel (100–200, mesh size) column chromatography using 13%–16% ethyl acetate in petroleum ether as the eluent to give 2 as a pale yellow solid (0.28 g, yield: 23%). 1H NMR (400 MHz, DMSO-d6) δ: 10.86 (s, 1H), 10.34 (s, 1H), 7.53 (s, 1H), 7.25 (s, 1H), 6.54 (d, J = 1.3 Hz, 1H), 2.40 (d, J = 1.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ: 190.65, 159.94, 156.66, 152.12, 146.09, 126.15, 124.84, 117.84, 115.52, 112.96, 18.44. HRMS (ESI): calculated for C11H8O4 [M − H+]− (m/z): 203.0350; found: 203.0358.
Synthesis of probe L. Rhodamine B hydrazide (1, 0.19 g, 0.42 mmol) and 7-formyl-6-hydroxy-4-methylcoumarin (2, 0.08 g, 0.42 mmol) were dissolved in anhydrous methanol (4 mL) and the solution was stirred for 1.5 h under refluxing conditions. After cooling to room temperature, the precipitate was filtered and washed three times with 10 mL cold methanol. The crude product was further purified through silica gel (100–200, mesh size) column chromatography using 10%–15% ethyl acetate in petroleum ether as eluent to give L as a pale yellow powder (0.14 g, yield: 53%). 1H NMR (400 MHz, CDCl3) δ: 12.23 (s, 1H), 9.62 (s, 1H), 7.99 (d, J = 7.0 Hz, 1H), 7.56–7.48 (m, 2H), 7.16 (d, t, J = 17.2, 9.1 Hz, 3H), 6.56 (d, J = 8.9 Hz, 2H), 6.48 (d, J = 2.5 Hz, 2H), 6.28 (d, d, J = 8.9, 2.5 Hz, 2H), 6.20 (s, 1H), 3.33 (q, J = 7.1 Hz, 8H), 2.22 (s, 3H), 1.15 (t, J = 7.0 Hz, 12H). 13C NMR (101 MHz, CDCl3) δ: 164.86, 160.16, 156.95, 153.23, 152.20, 151.69, 149.32, 148.17, 148.11, 134.01, 128.72, 128.24, 127.65, 123.93, 123.62, 122.21, 120.82, 118.76, 117.97, 113.05, 108.48, 104.72, 98.31, 66.21, 44.45, 25.39, 12.62. HRMS (ESI): calculated for C39H38N4O5 [M + H+]+ (m/z): 643.2915; found: 643.2921. FTIR (KBr) ν: 3445(–OH), 1727, 1715 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1616 (CN).
O), 1616 (CN).
Preparation of L–Cu2+ complex
CuCl2·2H2O (0.08 g, 0.45 mmol) was added to a stirred solution of receptor L (0.19 g, 0.30 mmol) in absolute ethanol. The solution was stirred at 50 °C and the reaction process was monitored by TLC. After the reaction was completed, the solvent was removed under reduced pressure and the solid complex was filtered, washed several times with deionized water and dried in vacuum to obtain a dark purple powder (0.17 g, yield: 76%). Elemental anal.: calculated for C39H39N4O6CuCl (%): C 61.74, H 5.18, N 7.38; found: C 61.89, H 5.32, N 6.82. HRMS (ESI): calculated for [L + Cu2+ − H+]+ (m/z): 704.2049, found: 704.2072. FTIR (KBr) ν: 3445 (–OH), 1716, 1699 (CO), 1590 (CN).
Results and discussion
Synthesis and structural characteristics of L
Receptor L was synthesized by the nucleophilic addition–condensation reaction of rhodamine B hydrazide (1) and 7-formyl-6-hydroxy-4-methylcoumarin (2) in absolute methanol (Scheme 1). L and the intermediate 2 were characterized by 1H NMR, 13C NMR and HRMS (ESI, Fig. S1–S6†).
Equilibration time
The equilibration time for complexation was evaluated between L and Cu2+ ion (Fig. S7†). No obvious absorbance variation of L (10 μM) at 562 nm was observed over a period of 15 min, indicating that the five-membered spirolactam structure of sensor L was stable. After the addition of Cu2+ ions, the absorbance intensity at 562 nm increased instantaneously and reached a maximum after 60 s. For further spectral measurements, a reaction time of 1 min was used to ensure that the reaction was completed.
Absorption spectrum of L in the presence of competitive metal ions
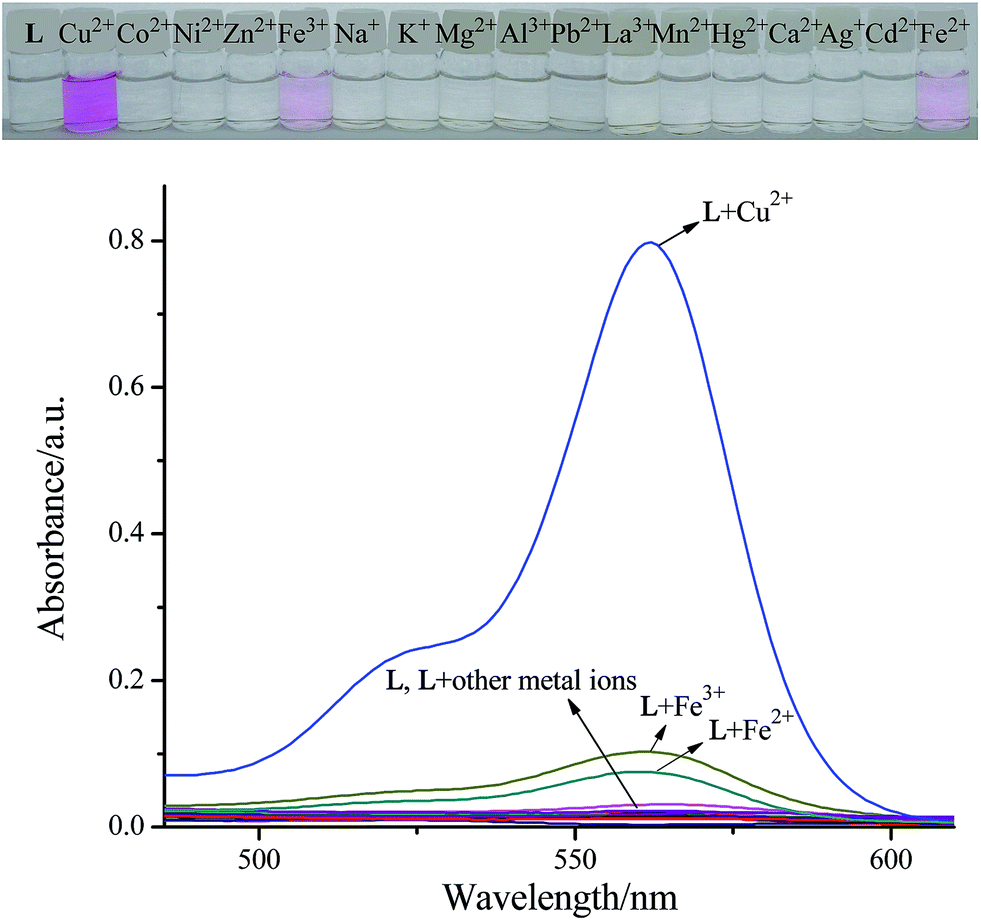
To gain an insight into the photochemical properties of L, absorption changes upon addition of various metal ions were performed in a DMSO–H2O (1:1, v/v) solution. As illustrated in Fig. 1, the characteristic absorption peak centered at 562 nm (266-fold absorbance enhancement in comparison with blank L) accompanied with remarkable color response from colorless to magenta was observed upon the addition of Cu2+. Among other tested metals, only Fe3+ and Fe2+ caused a slight color change from colorless to light pink, and a corresponding absorption peak appeared at 562 nm as expected, similar to that observed with Cu2+, whereas Na+, K+, Co2+, Ni2+, Zn2+, Mg2+, Al3+, Ca2+, Pb2+, Mn2+, Hg2+, Ag+, La3+ and Cd2+ exerted either little or no disturbance on the UV-Vis spectra of L. However, these changes induced by Fe3+ or Fe2+ were distinctly less in magnitude as compared to the changes observed with Cu2+ due to its low binding affinity to L, which indicates that composite L can serve as a potential chemosensor for the naked eye detection of Cu2+ in an aqueous medium.
 |
| | Fig. 1 Naked-eye detectable color changes and UV-Vis absorption spectra of L (10 μM, in DMSO) in a DMSO/H2O (1:1, v/v) solution upon addition of various metal ions (10 μM, in H2O). | |
UV-Vis titration experiment
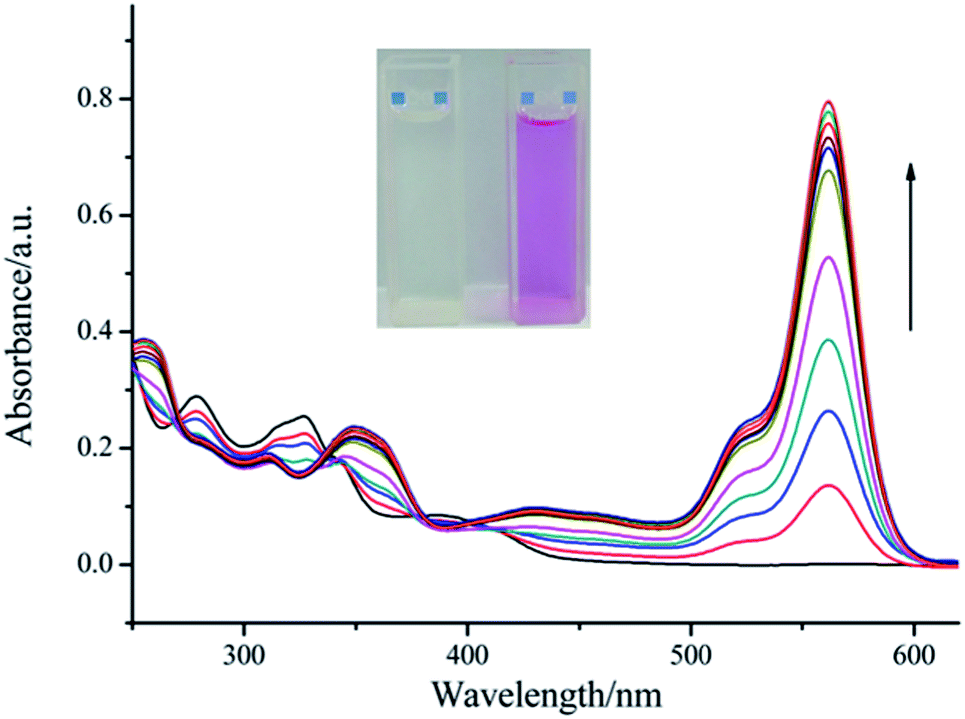
To further study the binding properties of L with Cu2+, we measured the absorption properties of L (10 μM) upon addition of an increasing concentration of Cu2+ (0–10 μM) (Fig. 2). With continuous addition of Cu2+ ions, a sharp absorption band centered at 562 nm emerged with increasing intensity, which induced an obvious color change from colorless to magenta. Moreover, three clear isosbestic points at 271 nm, 337 nm and 380 nm were observed, which indicate the formation of only one visible active copper complex. Furthermore, a linear dependence of the absorbance at 562 nm as a function of Cu2+ concentration was observed (Fig. S8†). These spectroscopic changes were characteristic of the spirolactam ring of rhodamine as the complexation process was accompanied by ring-opening. For all concentrations of Cu2+ ions above 10 μM, the intensity of absorption at 562 nm reached saturation, which suggested the formation of a 1:1 complex. The stoichiometry between L and Cu2+ was confirmed by utilizing the Job's method (Fig. 3). High resolution mass spectrometry (Fig. S9†) provided further support for the formation of the 1:1 complex (m/z calculated for [L + Cu2+ − H+]+ = 704.2049, m/z observed = 704.2072). On the basis of non-linear fitting of the titration curve of a 1:1 binding model, the association constant of the Cu2+–L complex was determined to be 5.23 × 104 M−1 (Fig. S10†). From the titration experiments, the detection limit for Cu2+ was calculated to be ∼28 nM, which was much lower than the WHO limit for Cu2+ (31.5 μM) in drinking water.20 This shows that our proposed method based on compound L has the potential to monitor the copper concentrations in water samples.
 |
| | Fig. 2 Titration curves of L (10 μM, in DMSO) in DMSO/H2O (1:1, v/v) solution upon addition of CuCl2·2H2O (0–10 μM, in H2O). Inset shows the color change of the solution before (left) and after (right) the addition of Cu2+. | |
 |
| | Fig. 3 Job's plot of L–Cu2+ complex in DMSO/H2O (1:1, v/v) solution. The total concentration of L and Cu2+ was 10 μM. The absorbance was monitored at 562 nm. | |
Tolerance of L to Cu2+ over other metal ions and anions
One basic requirement of an ion-selective chemosensor is its target selectivity over other competitive substrates. The absorbance response of L was highly selective for Cu2+ over biologically and environmentally relevant analytes (Fig. 4). No significant difference in the response of the L–Cu2+ system in the absence and presence of the interfering metal ions (5 equivalents) was observed, except for Hg2+. To clearly understand the interference of Hg2+ on the optical response of L to Cu2+, a UV-Vis titration experiment was carried out (Fig. S11†). Upon gradual addition of Hg2+ (0–60 μM) to a solution of L–Cu2+ complex (10 μM), a slight loss of color was observed. Moreover, the absorption band at 562 nm decreased with distinct isosbestic points at 333 nm, 372 nm and 413 nm, indicating the presence of UV-active species in equilibrium. For all concentrations of Hg2+ ions above 50 μM (i.e., 60 μM, 6 equiv.; 100 μM, 10 equiv.), the intensity of absorption at 562 nm almost reached saturation (Fig. S11–S12†). As a result, Hg2+ had a negative effect on the response of Cu2+, probably because of the strong affinity of Hg2+ to L for detaching Cu2+ from the L–Cu2+ complex15g, and the existing L–Hg2+ complex was colorless, which escapes visual inspection and hence has no spectral response was observed. However, the competitive coordination was removed and generally reached a balance when an excess of Hg2+ ions were added into the L–Cu2+ system. The effect of different organic and inorganic anions was also investigated under the same conditions. Evidently, the coexistence of these anions did not show any distinct influence on the recognition process of Cu2+ by L. To determine the effect of counter anions in the sensing behavior of receptor L, the response of L with Cu2+ from different copper salts such as copper chloride, copper sulfate, copper nitrate and copper acetate were investigated, as shown in Fig. S13.† When 10 μM of L encountered 1 equiv. of Cu2+, the response of L with Cu2+ from copper chloride was almost in accordance with Cu2+ from other copper salts. These results clearly demonstrated that receptor L can function as a highly selective/anti-disturbance sensor for Cu2+ and could be used for the practical detection of Cu2+ in water samples.
 |
| | Fig. 4 Absorbance intensity change profiles of L (10 μM, in DMSO) in the presence of Cu2+ (10 μM, in H2O) or Cu2+ (10 μM, in H2O) with other metal ions or anions (Mn+, Mn−, 50 μM, in H2O) in a DMSO/H2O (1:1, v/v) solution. (1) Blank; (2) Cu2+; (3) Cu2+ + Co2+; (4) Cu2+ + Ni2+; (5) Cu2+ + Zn2+; (6) Cu2+ + Fe3+; Cu2+ + CH3COO−; (7) Cu2+ + Na+; Cu2+ + F−; (8) Cu2+ + Mg2+; Cu2+ + Cl−; (9) Cu2+ + Al3+; Cu2+ + Br−; (10) Cu2+ + Pb2+; Cu2+ + I−; (11) Cu2+ + La3+; Cu2+ + HPO42−; (12) Cu2+ + Mn2+; Cu2+ + H2PO4−; (13) Cu2+ + Hg2+; Cu2+ + S2O32−; (14) Cu2+ + Ca2+; Cu2+ + CO32−; (15) Cu2+ + Ag+; Cu2+ + HCO3−; (16) Cu2+ + Cd2+; Cu2+ + SO42−; (17) Cu2+ + K+; Cu2+ + HSO4−; (18) Cu2+ + Fe2+; Cu2+ + NO3−. The absorbance was monitored at 562 nm. | |
Proposed mechanism
To obtain a clear understanding of the structure of the L–Cu2+ complex, FTIR measurements were primarily employed (Fig. 5). The characteristic stretching frequencies of L at 1727 cm−1 and 1616 cm−1, corresponding to ν (CO) (the rhodamine unit) and ν (CN), respectively, almost disappeared completely in the spectra of the L–Cu2+ complex. Moreover, the absorption peak of ν (–OH) at 3445 cm−1 decreased conspicuously. These spectroscopic changes are strong evidence that the spirolactam CO, CN and –OH groups participated in Cu2+ coordination.
 |
| | Fig. 5 (a) The whole FTIR spectra of L and L–Cu2+ complex. (b) The ∼1700 region of spectra of L and L–Cu2+ complex. | |
A 1H NMR titration experiment was further carried out in DMSO-d6. As shown in Fig. S14,† upon the addition of 1 equiv. of Cu2+ ions, reduction in the intensity of the hydroxyl group (–OH, δ = 10.43 ppm) accompanied with a slight up field chemical shift (δ = 0.03 ppm) was observed, indicating deprotonation as a result of interaction with Cu2+.
To rule out the possibility that the absorbance changes observed were not due to a chemical reaction (i.e., chemodosimeter21), the reversible binding of Cu2+ and the sensor was established. The reversibility experiment (Fig. S15†) revealed that the 266-fold absorbance enhancement of L caused by the addition of 1 equiv. of Cu2+ ions can be removed completely by adding 1.5 equiv. of EDTA–2Na, leading to the reconstitution of the spirolactam ring in the rhodamine moiety and hence the loss of absorbance at 562 nm.
On the basis of the combined spectroscopic information and previously reported literature15(c),22, a possible Cu2+-induced deprotonation mechanism and coordination mode of receptor L with Cu2+ was proposed, as shown in Scheme 2.
 |
| | Scheme 2 Proposed mechanism for the identification of Cu2+ triggered by L. | |
Practical application and recreational test
To investigate the preliminary application of chemosensor L, test strips were facilely prepared by immersing normal filter papers into a saturated DMSO solution of L and then drying in vacuum. These test strips were applied for sensing different Cu2+ concentrations, exhibiting colorimetric changes differentiable by the naked eye. As depicted in Fig. 6, the well-marked red color of the test strips intensified from 0 to 1.0 × 10−5 M, 1.0 × 10−4 M, 1.0 × 10−3 M, 1.0 × 10−2 M and 1.0 × 10−1 M and show that the discernible concentration of Cu2+ can be as low as 1.0 × 10−4 M. These results indicated that receptor L could serve as a good candidate for conveniently detecting Cu2+ in pure water samples.
 |
| | Fig. 6 Images of the test strips coated with L for colorimetric detecting Cu2+ ion in an aqueous solution with different concentrations. Left to right: 0, 1 × 10−1 M, 1 × 10−2 M, 1 × 10−3 M, 1 × 10−4 M and 1 × 10−5 M. | |
Taking advantage of the distinct abovementioned color change, a recreational test was then carried out using an A4 paper (Fig. 7). No obvious color change was observed when “ ” gaps were filled with Cu2+ (1.0 × 10−2 M). Interestingly, it would turn into magenta immediately after adding the saturated DMSO solution of L dropwise.
” gaps were filled with Cu2+ (1.0 × 10−2 M). Interestingly, it would turn into magenta immediately after adding the saturated DMSO solution of L dropwise.
 |
| | Fig. 7 Colorimetric response of Cu2+ ion in the absence or presence of L. (Top) none, (middle) Cu2+, (bottom) Cu2+ + L. | |
Conclusions
In summary, we developed a new chemosensor L as a colorimetric probe for the sensitive detection of Cu2+ ion with a short response time in a semi-aqueous medium. The complex exhibited high selectivity for Cu2+ ion over a panel of other metal ions. Importantly, the detection limit (28 nM) of L for Cu2+ falls sufficiently below the limit criterion of drinking water (31.5 μM). Due to the colorimetric response of L to Cu(II), test strips containing L were fabricated as a naked-eye indicator for Cu2+ ion in pure water samples, a recreational test was also achieved. Based on this simple, rapid and cost-effective method, we believe that receptor L will be an excellent prototype for the development of a novel colorimetric Cu2+-chemosensor.
Acknowledgements
The authors would like to thank the National Natural Science Foundation of China (Grant No. 21261008, 21471069, 1601310046) and the International Science and Technology Cooperation Projects of Hainan Province (Grant No. KJHZ2014-05).
Notes and references
-
(a) X. Chen, X. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783 RSC;
(b) X. Chen, T. Pradhan, F. Wang, J. S. Kim and J. Yoon, Chem. Rev., 2012, 112, 1910 CrossRef CAS PubMed;
(c) Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568 RSC;
(d) M. E. Moragues, R. Martínez-Máñez and F. Sancenón, Chem. Soc. Rev., 2011, 40, 2593 RSC;
(e) L. Gao, L. Li, X. Wang, P. Wu, Y. Cao, B. Liang, X. Li, Y. Lin, Y. Lu and X. Guo, Chem. Sci., 2015, 6, 2469 RSC.
-
(a) P. Verwilst, K. Sunwoo and J. S. Kim, Chem. Commun., 2015, 51, 5556 RSC;
(b) S. R. Patil, J. P. Nandre, P. A. Patil, S. K. Sahoo, M. Devi, C. P. Pradeep, F. Yu, L. Chen, C. Redshaw and U. D. Patil, RSC Adv., 2015, 5, 21464 RSC;
(c) J. Li, Y. Zeng, Q. Hu, X. Yu, J. Guo and Z. Pan, Dalton Trans., 2012, 41, 3623 RSC.
-
(a) G. J. Brewer, Curr. Opin. Chem. Biol., 2003, 7, 207 CrossRef CAS;
(b) S. Hu, J. Song, F. Zhao, X. Meng and G. Wu, Sens. Actuators, B, 2015, 215, 241 CrossRef CAS PubMed.
- R. B. Jonas, Appl. Environ. Microbiol., 1989, 55, 43 CAS.
-
(a) G. P. C. Rao, K. Seshaiah, Y. K. Rao and M. C. Wang, J. Agric. Food Chem., 2006, 54, 2868 CrossRef CAS PubMed;
(b) T. Kato, S. Nakamur and M. Mirita, Anal. Sci., 1990, 6, 623 CrossRef CAS.
- I. Karadjova, B. Izgi and S. Gucer, Spectrochim. Acta, Part B, 2002, 57, 581 CrossRef.
- T. Poursaberi, L. Hajiagha-Babaei, M. Yousefi, S. Rouhani, M. Shamsipur, M. Kargar-Razi, A. Moghimi, H. Aghabozorg and M. R. Ganjali, Electroanalysis, 2001, 13, 1513 CrossRef CAS.
- S. Hong, T. Kang, J. Moon, S. Oh and J. Yi, Colloids Surf., A, 2007, 292, 264 CrossRef CAS PubMed.
- K. M. Gattas-Asfura and R. M. Leblanc, Chem. Commun., 2003, 2684 RSC.
-
(a) F. J. Huo, C. X. Yin, Y. T. Yang, J. Su, J. B. Chao and D. S. Liu, Anal. Chem., 2012, 84, 2219 CrossRef CAS PubMed;
(b) M. Cui, Q. Liu, Q. Fei, Y. Fei, Y. Liu, H. Shan, G. Feng and Y. F. Huan, Anal. Methods, 2015, 7, 4252 RSC.
-
(a) S. Sun, B. Qiao, N. Jiang, J. Wang, S. Zhang and X. Peng, Org. Lett., 2014, 16, 1132 CrossRef CAS PubMed;
(b) J. Yin, Y. Kwon, D. Kim, D. Lee, G. Kim, Y. Hu, J.-H. Ryu and J. Yoon, J. Am. Chem. Soc., 2014, 136, 5351 CrossRef CAS PubMed;
(c) S. Khatua and M. Schmittel, Org. Lett., 2013, 15, 4422 CrossRef CAS PubMed;
(d) T. Anand, G. Sivaraman, P. Anandh, D. Chellappa and S. Govindarajan, Tetrahedron Lett., 2014, 55, 671 CrossRef CAS PubMed;
(e) J. F. Zhang, Y. Zhou, J. Yoon, Y. Kim, S. J. Kim and J. S. Kim, Org. Lett., 2010, 12, 3852 CrossRef CAS PubMed;
(f) J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973 CrossRef CAS PubMed;
(g) Y. Zhang, Z. Shi, L. Yang, X. Tang, Y. An, Z. Ju and W. Li, Inorg. Chem. Commun., 2014, 39, 86 CrossRef PubMed.
-
(a) R. Martínez, A. Espinosa, A. Tárraga and P. Molina, Tetrahedron, 2008, 64, 2184 CrossRef PubMed;
(b) G. Hennrich, W. Walter, U. Resch-Genger and H. Sonnenschein, Inorg. Chem., 2001, 40, 641 CrossRef CAS;
(c) V. Chandrasekhar, P. Bag and M. D. Pandey, Tetrahedron, 2009, 65, 9876 CrossRef CAS PubMed.
-
(a) W. Lin, L. Yuan, W. Tan, J. Feng and L. Long, Chem.–Eur. J., 2009, 15, 1030 CrossRef CAS PubMed;
(b) Z. Jiang, L. Tang, F. Shao, G. Zheng and P. Lu, Sens. Actuators, B, 2008, 134, 414 CrossRef CAS PubMed;
(c) H. Li, Z. Yang and D. Qin, Inorg. Chem. Commun., 2009, 12, 494 CrossRef CAS PubMed;
(d) H. S. Jung, P. S. Kwon, J. W. Lee, J. I. Kim, C. S. Hong, J. W. Kim, S. Yan, J. Y. Lee, J. H. Lee, T. Joo and J. S. Kim, J. Am. Chem. Soc., 2009, 131, 2008 CrossRef CAS PubMed;
(e) S. Kaur and S. Kumar, Tetrahedron Lett., 2004, 45, 5081 CrossRef CAS PubMed.
-
(a) J. Shao, Y. Qiao, H. Lin and H. Lin, Spectrochim. Acta, Part A, 2009, 71, 1736 CrossRef PubMed;
(b) N. Kaur and S. Kumar, Tetrahedron, 2011, 67, 9233 CrossRef CAS PubMed;
(c) Y. Li, Y. Duan, J. Zheng, J. Li, W. Zhao, S. Yang and R. Yang, Anal. Chem., 2013, 85, 11456 CrossRef CAS PubMed;
(d) Y. J. Na, G. J. Park, H. Y. Jo, S. A. Lee and C. Kim, New J. Chem., 2014, 38, 5769 RSC;
(e) J. Zhou, D. Liu, Y. He, X. Kong, Z. Zhang, Y. Ren, Y. Long, R. Huang and L. Zheng, Dalton Trans., 2014, 43, 11579 RSC.
-
(a) P. Kaur, S. Kaur and K. Singh, Org. Biomol. Chem., 2012, 10, 1497 RSC;
(b) M. Wang, K. H. Leung, S. Lin, D. S. H. Chan, D. W. J. Kwong, C. H. Leung and D. L. Ma, Sci. Rep., 2014, 4, 6794 CrossRef CAS PubMed;
(c) X. Xu, W. L. Daniel, W. Wei and C. A. Mirkin, Small, 2010, 6, 623 CrossRef CAS PubMed;
(d) P. Kaur, H. Kaur and K. Singh, RSC Adv., 2013, 3, 64 RSC;
(e) T. G. Jo, Y. J. Na, J. J. Lee, M. M. Lee, S. Y. Lee and C. Kim, New J. Chem., 2015, 39, 2580 RSC;
(f) Z. Xu, L. Zhang, R. Guo, T. Xiang, C. Wu, Z. Zheng and F. Yang, Sens. Actuators, B, 2011, 156, 546 CrossRef CAS PubMed;
(g) R. Sheng, P. Wang, Y. Gao, Y. Wu, W. Liu, J. Ma, H. Li and S. Wu, Org. Lett., 2008, 10, 5015 CrossRef CAS PubMed.
-
(a) J. Liu, H. Wang and X. Yan, Analyst, 2011, 136, 3904 RSC;
(b) J. Y. Noh, G. J. Park, Y. J. Na, H. Y. Jo, S. A. Lee and C. Kim, Dalton Trans., 2014, 43, 5652 RSC;
(c) X. Xie, X. Chen, B. Li and L. Zhang, Dyes Pigm., 2013, 98, 422 CrossRef CAS PubMed;
(d) H. Kim, Y. J. Na, E. J. Song, K. B. Kim, J. M. Bae and C. Kim, RSC Adv., 2014, 4, 22463 RSC;
(e) P. Kaur, D. Sareen and K. Singh, Talanta, 2011, 83, 1695 CrossRef CAS PubMed.
- B. K. Datta, D. Thiyagarajan, S. Samanta, A. Ramesh and G. Das, Org. Biomol. Chem., 2014, 12, 4975 CAS.
- M. Zhu, M. Yuan, X. Liu, J. Xu, J. Lv, C. Huang, H. Liu, Y. Li, S. Wang and D. Zhu, Org. Lett., 2008, 10, 1481 CrossRef CAS PubMed.
- J. Zhang, B. Li, L. Zhang and H. Jiang, Chem. Commun., 2012, 48, 4860 RSC.
- WHO, WHO Guidelines Values for Chemicals that are of Health Significance in Drinking Water, Guidelines for Drinking Water Quality, Geneva, 3rd edn, 2008 Search PubMed.
- M. H. Lee, B.-K. Cho, J. Yoon and J. S. Kim, Org. Lett., 2007, 9, 4515 CrossRef CAS PubMed.
-
(a) S. Goswamia, D. Sena, A. K. Dasa and N. K. Dasa, Sens. Actuators, B, 2013, 183, 518 CrossRef PubMed;
(b) W. Y. Liu, H. Y. Li, H. S. Lv, B. X. Zhao and J. Y. Miao, Spectrochim. Acta, Part A, 2012, 95, 658 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, ESI-MS data of compounds 2 and L, ESI-MS data of L–Cu2+ complex, 1H NMR titration data and additional spectroscopic studies. See DOI: 10.1039/c5ra12407g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 













 ” gaps were filled with Cu2+ (1.0 × 10−2 M). Interestingly, it would turn into magenta immediately after adding the saturated DMSO solution of L dropwise.
” gaps were filled with Cu2+ (1.0 × 10−2 M). Interestingly, it would turn into magenta immediately after adding the saturated DMSO solution of L dropwise.