
Enhanced actuated strain of titanium dioxide/nitrile-butadiene rubber composite by the biomimetic method†
Abstract
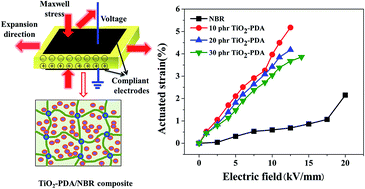
In order to improve compatibility between the dielectric filler and polymeric matrix, we used bio-inspired polydopamine (PDA) to modify titanium dioxide (TiO2) nano-particles. The PDA coated TiO2 (TiO2-PDA) nano-particles were incorporated into nitrile-butadiene rubber (NBR) which contains a large amount polar groups to obtain a dielectric elastomer composite with a large actuated strain under a low electric field. The relatively soft insulating PDA layer on the TiO2 nano-particles led to the composites filled with TiO2-PDA nano-particles displaying better filler dispersion, much lower elastic modulus, lower dielectric loss, and higher electric breakdown field compared with the composites filled with pristine TiO2 nano-particles, resulting in a high electromechanical sensitivity (β). At last, an actuated strain of 5.2% at a relatively safe electric field of 12.5 kV mm−1 without any pre-strains was obtained by the 10 phr TiO2-PDA/NBR composite, a 140% increase in actuated strain compared with the actuated strain (0.69%) of pure NBR at 20 kV mm−1 without any pre-strains. This biomimetic method is simple, efficient, nontoxic, and easy to control, which can be used in other dielectric fillers to improve electromechanical properties of dielectric elastomers.
 Please wait while we load your content...
Please wait while we load your content...

