DOI:
10.1039/C5RA12217A
(Paper)
RSC Adv., 2015,
5, 64806-64813
Role of polyethyleneimine as an additive in cyanide-free electrolytes for gold electrodeposition†
Received
24th June 2015
, Accepted 23rd July 2015
First published on 23rd July 2015
Abstract
The influence of polyethyleneimine (PEI) on the electrochemical behavior of cyanide-free electrolytes with 5,5-dimethylhydantoin (DMH) as a complexing agent was investigated using cyclic voltammetry (CV), chronoamperometry, and cathodic polarization measurements. The gold electrodeposition displayed three-dimensional (3-D) progressive nucleation in both the absence and presence of PEI according to the Scharifer and Hills (SH) nucleation model. With the addition of PEI, the cathodic overpotential showed an immense negative shift, and the limiting diffusion current density and the diffusion coefficient decreased. Gold electrodeposits were characterized by SEM and AFM for the micromorphology, XRD for the crystal structure, and XPS for the purity. There was preferential growth of gold crystals along the (111) crystal face in both the presence and absence of PEI, but overall growth was inhibited in the presence of PEI. The quality of the gold electrodeposit was significantly improved in the presence of PEI, with a bright golden color and high purity, smooth and compact morphology.
1. Introduction
Owing to its excellent physicochemical properties and elegant appearance, gold electrodeposits are widely used for decorative and functional applications.1–4 Gold deposits are applied in electronics as a functional coating to improve physical properties such as conductivity, corrosion and wear resistance.5 With the rapid development of interconnection and nanosensor fabrication, nanocrystalline materials with fine grains are urgently needed to meet the increasing demand of nano- and microdevices.6,7 Due to its excellent electrical and thermal conductivity, chemical stability, high ductility, and wear resistance, nanocrystalline gold electrodeposits are an ideal material in several fields8–11 such as microelectronics,12 microsystem technologies,13 biotechnology,14–16 surface modification,17–19 and basic scientific research.20
In order to replace the application of hazardous cyanide in gold electrodeposition, a number of cyanide-free gold electroplating electrolytes have been investigated over recent years. Nowadays, the most commonly used cyanide-free gold electroplating electrolytes use sulfite as the complexing agent.21,22 However, these sulfite-based gold electroplating electrolytes still suffer from problems of instability.23 5,5-dimethylhydantoin (DMH), a promising environmentally friendly complexing agent, can replace cyanide due to its good coordinating capability with several metal ions such as gold, silver, and copper ions.24–27 More attention has been paid to DMH-based gold electroplating electrolytes because it is simple, stable, and environmentally friendly. H NMR spectra of the gold electroplating electrolytes were employed to reveal that strong coordination bonding of DMH− to Au(III) was present, and that [Au(DMH)4]− was the quantitative complexation structure.24 DMH was selected as a complexing agent for gold electroplating in our study,28 and the gold electrodeposit quality was preliminarily studied. The preliminary gold electrodeposit obtained from this DMH-based electrolyte was not sufficiently bright, and had a rough, uncompact surface. Additives were needed to improve the appearance of the gold electrodeposit. The importance of using additives in gold electroplating electrolytes has been recognized, but there are few investigations about the influence of these additives on the gold electrodeposition process.22,26,29 During the research of cyanide-free gold electroplating electrolytes, investigation of the electroplating mechanism of gold is essential for further improvement of gold electroplating electrolytes and the quality of the gold electrodeposit.
In this work, polyethyleneimine (PEI), an organic molecule rich in positively charged amino groups,15 was selected as the additive in DMH-based gold electroplating electrolytes. The influence of PEI on the gold electrodeposition process in DMH-based electrolytes was investigated by CV, chronoamperometry, and cathodic polarization. The performance of the gold electrodeposit was evaluated by scanning electron microscope (SEM), atomic force microscopy (AFM), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS).
2. Experimental procedure
The base gold electroplating electrolytes were prepared with 0.025 mol L−1 HAuCl4, 0.3 mol L−1 DMH, and 0.36 mol L−1 K2CO3 in deionized water. The pH of all the electrolytes was maintained at 10. PEI used in gold electroplating electrolytes as additive is 30 mg L−1, which was optimized by electrochemical measurement and material characterization through the influence of PEI to the gold nucleation process and the morphology of gold electrodeposits, as displayed in Fig. S1 and S2 (ESI†). All of the reagents used in this work were analytical grade. The gold electroplating electrolyte in the presence of 30 mg L−1 PEI also had broad operating current density and good throwing power, which studied by Hull cell (Fig. S3, ESI†) and Haring-Blum cell (ESI†), respectively.
All electrochemical measurements were performed in a typical three-electrode cell using an electrochemical workstation at 318 K. A platinum foil and a saturated calomel electrode (SCE) were employed as the counter electrode (CE) and the reference electrode (RE), respectively. A platinum rotating disk electrode (Pt-RDE) with a diameter of 4 mm was employed as the working electrode (WE) for the cathodic measurements, and a glassy carbon electrode (GCE) with a diameter of 3 mm was used for CV and chronoamperometry measurements. The solution viscosity was measured by a digital viscometer.
Gold electrodeposits were deposited on copper sheet, which was firstly coated with a nickel deposit, an important diffusion barrier layer. The sample was face to face with the anode, and the distance between them was about 5 cm in the experiments. As there is hydrogen evolution reaction during the gold electrodeposition, bath agitation is important to enhance the mass transport and improve the macroscopic and microscopic performance of the gold electrodeposit, as displayed in Fig. S4 and S5 (ESI†). Gold electrodeposit characteristics were evaluated by SEM, AFM, XRD, and XPS to study the surface morphologies, crystal structures, and purity. Gold electrodeposits were all prepared under galvanostatic conditions (8 mA cm−2) with mild agitation at 318 K, and the current efficiency of the gold electroplating electrolytes were around 97% and 92% in the absence and presence of 30 mg L−1 PEI, respectively. The surface morphologies and microstructure of the gold electrodeposits were studied by field emission scanning electron microscopy (FE-SEM, Hitachi SU8000). An atomic force microscope (AFM) was employed to study the surface roughness of the gold electrodeposits. The AFM analysis was carried out with a dimension icon (Bruker) working in contact mode with silicon nitride cantilevers. X-Ray Diffraction (XRD) analysis was carried out on a D/max-3C X-ray diffractometer at a scanning rate of 0.02° s−1 with Cu Kα radiation. The elemental analysis of the gold electrodeposit was performed to analyze the purity of the deposit by X-ray photoelectron spectroscopy (XPS) on a PHI 5700 ESCA System (Physic Electronics, USA), with an excitation source of Al Kα radiation (photoelectron energy of 1486.6 eV) and hemispherical precision electron energy analyzer.
3. Results and discussion
3.1 Cyclic voltammograms
CV on a GCE was employed to study the electrochemical behavior of Au(III) of DMH-based gold electroplating electrolytes in the absence or presence of PEI. With a scan rate of 10 mV s−1, the CV began scanning at the open circuit potential (OCP) in the negative direction, along a range of −1.6 to 1.55 V and −1.15 to 1.5 V in the absence or presence of PEI, respectively, as shown in Fig. 1. As seen, both of these voltammograms consist of a cathodic peak, a hydrogen evolution area, and an oxygen evolution area.
 |
| | Fig. 1 Cyclic voltammograms with a scan rate of 10 mV s−1 on the GCE of the DMH-based electrolytes (a) in the absence of PEI and (b) in the presence of 30 mg L−1 PEI. | |
During the initial scan along the negative direction, the cyclic voltammogram of the base electrolyte rises at around −0.51 V, inducing the reduction of [Au(DMH)4]− to Au and the nucleation of Au on the GCE surface. With the addition of PEI into the base electrolyte, the rise occurs at around −0.62 V, 110 mV more negative than that of the base electrolyte. Furthermore, the cyclic voltammogram of the base electrolyte reveals a cathodic current density peak of −5.83 mA cm−2 at −0.659 V, much lower than that of the electrolyte in the presence of PEI, which is −13.77 mA cm−2 at −0.753 V. The onset potentials corresponding to hydrogen evolution are around −1.33 V without PEI, and −0.97 V for the electrolyte with PEI. In addition, both of the cyclic voltammograms show a “hysteresis loop”,26 which is characteristic of three-dimensional (3-D) nucleation and growth of gold nuclei on the GCE surface. These results indicate that the onset potential of gold electrodeposition from the electrolyte with PEI has quite more negative nucleation overpotential and a less negative overpotential for hydrogen evolution than that of the base electrolyte. In both electrolytes with or without PEI, the area of the oxygen evolution reaction occurs when the scanning potential is beyond 1.03 V, and no anodic peak corresponding to anodic dissolution is observed on the anodic sides of the cyclic voltammograms. Moreover, both the gold electroplating electrolytes in the absence and presence of PEI were slightly influenced by time and had good chemistry stability, as shown in Fig. S6 (ESI†).
3.2 Nucleation mechanism
Chronoamperometry30 was used to investigate the initial nucleation stage of the gold electrodeposition process influenced by PEI. Potentiostatic current transients on the GCE from electrolytes in the absence or presence of PEI are presented in Fig. 2. The applied potentials (Eap) were chosen to be more negative than the cathodic peaks of the cyclic voltammograms to ensure that the electrochemical reactions were under diffusion control. As shown in Fig. 2, all the curves obtained from the gold electroplating electrolytes, in the absence or presence of PEI, show a current decrease at the very beginning, which results from double-layer charging. After that, the current densities increase due to simultaneous gold nuclei growth and an increasing number of nuclei. With the growth of gold nuclei, the diffusion zones around them expand and finally overlap with the diffusion zones of neighboring nuclei, while the current density reaches jm (the maximum of the current density j). An increase in jm and a decrease in tm (measured time corresponding to jm) is observed as Eap is set to more negative values. The existence of a current density peak in the current transients indicates that the charge transfer process controls the reaction kinetics at early stages of gold electrodeposition, indicating that there is a nucleation stage preceding the growth of the gold deposit. Contrasting Fig. 2(a) with (b), tm of the current transients obtained from the base electrolyte are smaller than those of the electrolyte in the presence of PEI at the same Eap (−0.76 V and −0.80 V), meaning it is more difficult to achieve charge transfer reaction control while using PEI. All the experimental current transients displayed in Fig. 2 show a tendency to reach the same limiting value after several seconds under different Eap, in either the absence or presence of PEI, demonstrating that the nucleation reaction reaches the diffusion controlled electrochemical process in either case.
 |
| | Fig. 2 Current transients at various applied potentials on the GCE of the DMH-based electrolytes (a) in the absence of PEI and (b) in the presence of 30 mg L−1 PEI. | |
The nucleation mechanism models proposed by Scharifer and Hills (SH)31,32 are famous and widely used in many reported theoretical models of nucleation mechanisms. These models provide a simple and fast way to classify experimental transients into two limiting cases, instantaneous or progressive nucleation, assuming both display 3-D growth of nuclei and are diffusion controlled over long periods of time. In the instantaneous nucleation process, all nuclei instantaneously form on the active electrode surface, while in the progressive nucleation process the number of nuclei increases with the extension of reaction time.
The SH model was employed to analyze the current transients measured in this work. All experimental current transients were transformed into non-dimensional plots of (j/jm)2 vs. (t/tm), and the plots were compared with theoretical current transients curves, as shown in Fig. 3(a) and (b). The SH models31 of 3-D instantaneous nucleation and progressive nucleation are shown in eqn (1) and (2), respectively:
| |
 | (1) |
| |
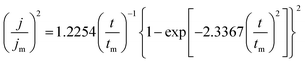 | (2) |
where
j and
t are the current and time, and
jm and
tm are the maximum current and the corresponding time, respectively. As displayed in
Fig. 3, compared to the theoretical curves for instantaneous and progressive nucleation, it is clear that the nucleation of the gold electroplating electrolyte in both the absence and presence of PEI displays progressive nucleation. The process of nucleation is not changed by a difference in
Eap, despite differences in
Eap affecting the ease at which gold nuclei are formed during the gold electrodeposition.
 |
| | Fig. 3 Non-dimensional plots of instantaneous and progressive nucleation models with three-dimensional nuclei growth and experimental curves of the DMH-based electrolytes (a) in the absence of PEI and (b) in the presence of 30 mg L−1 PEI. | |
To further study the gold nucleation and growth behavior in both electrolytes with and without PEI, SEM images (Fig. 4) were taken after electrodeposition of gold on GCE electrodes at Eap = −0.80 V for 1, 5, and 10 s. As shown in Fig. 4, as time increases both the gold nuclei growth and number of nuclei increase simultaneously, and the gold nucleation processes show progressive nucleation in both the electrolyte with and without PEI. Moreover, it can be clearly observed that the gold nuclei obtained from the electrolyte in the presence of PEI are much smaller than those obtained from the base electrolyte.
 |
| | Fig. 4 SEM images of gold electrodeposited on GCE electrodes at −0.80 V obtained from the DMH-based electrolytes in the absence of PEI at (A) 1 s, (B) 5 s, (C) 10 s, and in the presence of 30 mg L−1 PEI at (a) 1 s, (b) 5 s, (c) 10 s. | |
3.3 Effect of additive in the electrolyte
Cathodic polarization measurements were performed at a scan rate of 1 mV s−1 with different rotational speeds, 400, 600, 800, 1000, and 1200 rpm on a Pt-RDE to analyze the limiting diffusion current density and the diffusion coefficient in order to study the influence of PEI on electrochemical behaviors in the DMH-based gold electroplating electrolytes. As shown in Fig. 5, the polarization curves from the electrolyte in the presence of PEI begin to rise at around −0.70 V, increasing sharply at around −0.80 V, contrasting to those of the base electrolyte which rise at around −0.45 V. This indicates that the cathodic polarization overpotentials are more negative in the presence of PEI, according to the results of CV. The sharp rise in the polarization curves likely results from the inhibitory effect of PEI on gold electrodeposition, by way of absorption on the substrate surface.
 |
| | Fig. 5 Cathodic polarization curves with a scan rate of 1 mV s−1 on the Pt-RDE at different rotational speeds, measuring the DMH-based electrolytes (a) in the absence of PEI and (b) in the presence of 30 mg L−1 PEI. | |
All the cathodic polarization curves of the electrolytes in both the absence and presence of PEI exhibit a platform corresponding to the limiting diffusion current densities (jd), occurring at around −0.95 V. Moreover, the limiting diffusion current density decreases in the presence of PEI, as displayed in Fig. 6. The jd in electrolytes both with and without PEI increase with an increase of rotational speed, which is characteristic of mass transfer control. According to the Levich equation, the diffusion coefficient can be calculated based eqn (3) and (4):
| |
 | (3) |
| |
 | (4) |
where
v is the viscosity of the solution,
ω is the rotation speed of the Pt-RDE, and
cB is the concentration of electroactive species in the electrolyte (0.025 mol L
−1 for electrolytes in both the absence and presence of PEI). The fitted
jd vs. ω1/2 plots, presented in
Fig. 6, possess a zero intercept and match well with Levich equation, indicating that the nucleation process is diffusion controlled at the limiting diffusion current density in the DMH-based gold electroplating electrolyte, both with and without PEI.
 |
| | Fig. 6 The limiting diffusion current densities (jd) versus ω1/2 of the DMH-based electrolytes (a) in the absence of PEI and (b) in the presence of 30 mg L−1 PEI. | |
The viscosities (v) of the DMH-based gold electroplating electrolytes in the absence or presence of PEI were measured, and D was calculated, as listed in Table 1. According to the results in Table 1 and v has no significant change with the addition of PEI, D was reduced slightly with the addition of PEI. It indicates that with the addition of PEI into the gold electroplating electrolyte, the ion transfer was inhibited.
Table 1 v and D of the DMH-based electrolytes in the absence and presence of PEI
| |
In the absence of PEI |
In the presence of PEI |
| v × 10−6/m2 s−1 |
1.07 |
1.08 |
| D × 10−6/cm2 s−1 |
6.8 ± 0.1 |
6.4 ± 0.1 |
3.4 Micromorphology of gold electrodeposits
Observed visually, the gold electrodeposits obtained from the electrolyte with PEI were bright gold, while the electrodeposits from the base electrolyte were brown. SEM images were employed to study the influence of PEI on the surface micromorphology of the gold electrodeposits, as shown in Fig. 7. A rough, loose morphology with big crystal particles was present in the gold electrodeposits obtained from the base electrolyte, while the gold electrodeposits obtained in the presence of PEI were smooth and compact with small crystal particles. This indicates that the quality of the gold electrodeposit is significantly improved with the addition of PEI. Both the gold electrodeposits obtained in the absence and presence of 30 mg L−1 PEI performed a close combination with the substrate, as displayed in Fig. S7 (ESI†).
 |
| | Fig. 7 SEM images of the gold electrodeposits obtained from the DMH-based electrolytes (a) in the absence of PEI and (b) in the presence of 30 mg L−1 PEI. | |
AFM images were utilized to study the influence of PEI on the 3-D morphology of the gold electrodeposits, as displayed in Fig. 8. Fig. 8(a) shows that the morphology of the gold electrodeposit is rough with big crystal particles on the surface, while Fig. 8(b) displays that the gold surface is smooth and compact with small crystal particles. The Ra, Rq, and Rmax in Fig. 8(a) and (b), checked by NanoScope Analysis, are displayed in Table 2, which illustrates that Ra, Rq, and Rmax of the gold electrodeposits obtained in the presence of PEI are all smaller than those obtained from the base electrolyte. Both the SEM and AFM results confirm that the gold electroplating electrolyte possesses excellent leveling capability in the presence of PEI.
 |
| | Fig. 8 AFM three-dimensional height images of the top view of the gold electrodeposits obtained from the DMH-based electrolytes (a) in the absence of PEI and (b) in the presence of 30 mg L−1 PEI, inset showed the AFM two-dimensional height images of the gold electrodeposits obtained in the absence and presence of PEI. | |
3.5 Crystallization analysis of gold electrodeposits
XRD patterns of the gold electrodeposits obtained in the absence or presence of PEI are shown in Fig. 9. There are five obvious peaks indexed to Au (111), (200), (220), (311), and (222) crystal faces at 2 theta (2θ) values of 38.2°, 44.4°, 64.6°, 77.6°, and 81.7°, respectively. The diffraction peak intensity of the (111) faces for gold electrodeposits obtained from both of the electrolytes with or without PEI is higher than that of any other peak, indicating that the reduction of Au(III) to Au occurred preferentially on Au (111) faces, as expected. There is a peak indexed to the Au (400) crystal face at a 2θ value of 98.1° for XRD patterns of the gold electrodeposit obtained from the base electrolyte, while there is no such obvious peak for the gold electrodeposit in the presence of PEI.
 |
| | Fig. 9 XRD patterns of (a) Au (JCPDS file: 04-0784) and the gold electrodeposits obtained from the DMH-based electrolytes (b) in the absence of PEI and (c) in the presence of 30 mg L−1 PEI. | |
3.6 Purity of gold electrodeposits
It is well known that the properties and quality of the gold deposit strongly depend on the morphology and purity of the gold deposit, as the electrical properties of the gold deposit can be affected by impurities. XPS analysis was employed to verify the purity of the gold electrodeposit to get a general idea about the components of the bulk gold and the gold surface. The gold electrodeposit may be made impure by the interfusing of DMH or PEI, which were used as the complexing agent and additive of the gold electroplating electrolyte, respectively. Fig. 10 displays the XPS spectra taken at the surface and at a 10 nm depth of the gold electrodeposit, which was rinsed with deionized water and dried with a gentle flow of air after being electrodeposited from the electrolyte in the presence of PEI.
 |
| | Fig. 10 XPS spectra at the surface and a 10 nm depth of gold electrodeposits obtained from the DMH-based electrolyte in the presence of 30 mg L−1 PEI, high resolution XPS spectra of (a) Au, (b) C, (c) N, and (d) O. | |
The XPS survey scan was taken over a wide binding energy region, from 1.200 to 1350.000 eV. The binding energy of the Au 4f7/2 peak at 84.0 eV (ref. 33 and 34) was used as an internal standard of the XPS survey scan for the electrodeposit at the surface and at a 10 nm depth, as the C 1s internal standard might be influenced by either PEI or other contaminants absorbed on the gold surface. As shown in Fig. 10, obvious signals for Au, C, N, and O are observed on the original gold surface, while almost no C, N, or O signals can be observed at a 10 nm depth of the gold electrodeposit, which indicates that neither DMH nor PEI are detected at a 10 nm interior depth of gold electrodeposit, as both DMH and PEI are nitrogen containing. Therefore, there was neither DMH nor PEI entrained into the gold electrodeposit and the purity of the gold electrodeposit was very high.
4. Conclusion
The influence of polyethyleneimine (PEI) on gold electrodeposition in DMH-based cyanide-free gold electroplating electrolytes was investigated through electrochemical measurements on GCE or Pt-RDE. CV measurements showed that the nucleation overpotential of Au(III) shifted negatively with the addition of PEI. And both the gold electroplating electrolytes in the absence and presence of PEI had good chemistry stability. The nucleation of gold electrodeposition in both the absence or presence of PEI displayed 3-D progressive nucleation according to SH nucleation modes. With an increasing measurement of time, both gold nuclei growth and the number of nuclei increased. The gold nuclei obtained from the electrolyte in the presence of PEI were much smaller than those obtained from the base electrolyte. The cathodic polarization curves on Pt-RDE confirmed the inhibitory effect of PEI on gold electrodeposition, which resulted in more negative nucleation overpotential. The limiting diffusion current density decreased in the presence of PEI, and the nucleation processes were diffusion controlled at the limiting diffusion current density in both the absence and presence of PEI. The diffusion coefficients were (6.8 ± 0.1) × 10−6 cm2 s−1 and (6.4 ± 0.1) × 10−6 cm2 s−1 in electrolytes without and with PEI, respectively, illustrating that the diffusion coefficient was slightly decreased with the addition of 30 mg L−1 PEI.
SEM and AFM images showed that the golden appearance of the gold electrodeposits obtained from the electrolyte with PEI were microscopically smooth and compact, with small and uniform crystal grains. These results further confirm the leveling capability of PEI on gold electrodeposition. The preferential growth orientation of gold crystal along the (111) crystal face was not changed in the presence of PEI as shown by XRD. In addition, the gold electrodeposit was very pure, as demonstrated by XPS results.
Acknowledgements
The authors are grateful for financial support from the State Key Laboratory of Urban Water Resource and Environment (Harbin Institute of Technology) (2015DX09). G. W. also acknowledges the financial support from the start-up fund of University at Buffalo, SUNY along with New York Center of Excellence of Materials Informatics.
References
- Z. Liu and A. C. West, Electrochim. Acta, 2011, 56, 3328–3333 CrossRef CAS PubMed.
- A. I. de Sá, S. Eugénio, S. Quaresma, C. M. Rangel and R. Vilar, Thin Solid Films, 2011, 519, 6278–6283 CrossRef PubMed.
- F. Mirkhalaf, K. Tammeveski and D. J. Schiffrin, Phys. Chem. Chem. Phys., 2009, 11, 3463–3471 RSC.
- T. Fujita, S. Nakamichi, S. Ioku, K. Maenaka and Y. Takayama, Sens. Actuators, A, 2007, 135, 50–57 CrossRef CAS PubMed.
- S. Dutta, M. Imran, A. Pandey, T. Saha, I. Yadav, R. Pal, K. K. Jain and R. Chatterjee, J. Mater. Sci.: Mater. Electron., 2014, 25, 382–389 CrossRef CAS.
- Z. Zeng, X. Long, H. Zhou, E. Guo, X. Wang and Z. Hu, Electrochim. Acta, 2015, 163, 107–115 CrossRef CAS PubMed.
- R. Jafari Jam, M. Heurlin, V. Jain, A. Kvennefors, M. Graczyk, I. Maximov, M. T. Borgström, H. Pettersson and L. Samuelson, Nano Lett., 2015, 15, 134–138 CrossRef CAS PubMed.
- S. Cherevko and C.-H. Chung, Electrochem. Commun., 2011, 13, 16–19 CrossRef CAS PubMed.
- R. K. Sarvestani and J. D. Williams, Electrochem. Solid-State Lett., 2010, 13, D37–D39 CrossRef CAS PubMed.
- A. J. Cobley, T. J. Mason, M. Alarjah, R. Ashayer and S. H. Mannan, Ultrason. Sonochem., 2011, 18, 37–41 CrossRef CAS PubMed.
- N. Togasaki, Y. Okinaka, T. Homma and T. Osaka, Electrochim. Acta, 2005, 51, 882–887 CrossRef CAS PubMed.
- T. A. Green, Gold Bull., 2007, 40, 105–114 CrossRef CAS.
- B. Yin, T. Jianhua, W. Jinfen, B. Chao and X. Shanhong, IET Nanobiotechnol., 2014, 8, 31–36 CrossRef.
- Y. Zhao, L. Z. Fan, J. L. Ren and B. Hong, J. Solid State Electrochem., 2014, 18, 1099–1109 CrossRef CAS.
- L. Lu, J. Wu, M. Li, T. Kang and S. Cheng, Microchim. Acta, 2015, 182, 233–239 CrossRef CAS.
- S. Saraf, C. J. Neal, S. Park, S. Das, S. Barkam, H. J. Cho and S. Seal, RSC Adv., 2015, 5, 46501–46508 RSC.
- Y. Bu and S. W. Lee, Microchim. Acta, 2015, 1–9 Search PubMed.
- P. Rameshkumar and R. Ramaraj, J. Electroanal. Chem., 2015, 741, 64–70 CrossRef CAS PubMed.
- Y. Wang, B. Song, J. Xu and S. Hu, Microchim. Acta, 2015, 182, 711–718 CrossRef CAS.
- S. Sobri, S. Roy, D. Aranyi, P. M. Nagy, K. Papp and E. Kalman, Surf. Interface Anal., 2008, 40, 834–843 CrossRef CAS PubMed.
- G. Baltrūnas, A. Valiūnienė, J. Vienožinskis, E. Gaidamauskas, T. Jankauskas and Ž. Margarian, J. Appl. Electrochem., 2008, 38, 1519–1526 CrossRef.
- J. Chen, Y. Fang, Q. Qiu, L. You, J. Song, G. Zhang, G. Chen and J. Sun, Green Chem., 2011, 13, 2339–2343 RSC.
- E. C. Estrine, S. Riemer, V. Venkatasamy, B. J. H. Stadler and I. Tabakovic, J. Electrochem. Soc., 2014, 161, D687–D696 CrossRef PubMed.
- K. Oyaizu, Y. Ohtani, A. Shiozawa, K. Sugawara, T. Saito and M. Yuasa, Inorg. Chem., 2005, 44, 6915–6917 CrossRef CAS PubMed.
- J. Zhang, A. Liu, X. Ren, J. Zhang, P. Yang and M. An, RSC Adv., 2014, 4, 38012–38026 RSC.
- X. Yang, M. An, Y. Zhang and L. Zhang, Electrochim. Acta, 2011, 58, 516–522 CrossRef CAS PubMed.
- A. Liu, X. Ren, B. Wang, J. Zhang, P. Yang, J. Zhang and M. An, RSC Adv., 2014, 4, 40930–40940 RSC.
- X. Ren, Y. Song, A. Liu, J. Zhang, P. Yang, J. Zhang and M. An, RSC Adv., 2015 10.1039/c5ra13140e.
- M. Hosseini and S. Ebrahimi, J. Electroanal. Chem., 2010, 645, 109–114 CrossRef CAS PubMed.
- S. Wu, Z. Yin, Q. He, G. Lu, Q. Yan and H. Zhang, J. Phys. Chem. C, 2011, 115, 15973–15979 CAS.
- B. Scharifker and G. Hills, Electrochim. Acta, 1983, 28, 879–889 CrossRef CAS.
- M. E. Hyde and R. G. Compton, J. Electroanal. Chem., 2003, 549, 1–12 CrossRef CAS.
- M. P. Seah, G. C. Smith and M. T. Anthony, Surf. Interface Anal., 1990, 15, 293–308 CrossRef CAS PubMed.
- I. Lindau, P. Pianetta, K. Y. Yu and W. E. Spicer, Phys. Rev. B: Solid State, 1976, 13, 492–495 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra12217a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 













