
Surfactant-free aqueous preparation from a star polymer of size-controlled nanoparticles with encapsulated functional molecules†
Abstract
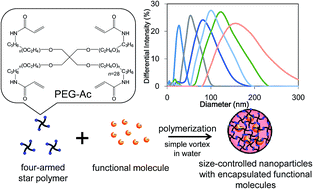
A simple method based on the vortex mixing of precursor chemicals in surfactant-free aqueous conditions has been developed for preparing nanoparticles that encapsulate functional molecules within a four-armed star polymer. Through investigation into the effects of altering such variables as the vortex intensity, chemical composition, temperature etc., it was found that the size of these nanoparticles could be controlled within a range of 20 to 200 nm. Furthermore, a variety of functional molecules could be encapsulated without changing the size of the nanoparticles, suggesting that this method has the potential to be used for a wide range of applications, such as medicine, industrial production and cosmetics.
 Please wait while we load your content...
Please wait while we load your content...

