
Phenomenon of current occurrence during the motion of a C60 fullerene on substrate-supported graphene
Abstract
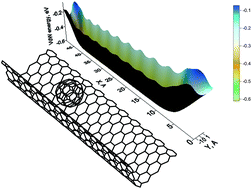
This paper studies the patterns of behavior of the fullerene C60 on graphene supported by an SiO2 substrate, taking into account the substrate topology and features of the electronic interaction between graphene and fullerene. It is found that the motion of the C60 molecule acquires a finite character when the substrate has a certain determined degree of curvature. We establish that such a character of motion occurs at a corrugation wavelength of 3.4 nm and wave-depth of 1.6 nm. The motion of the fullerene becomes more precise with deviations of tenths of an angstrom under an external electric field, which allows the motion of the fullerene to be manipulated along the trough. During the investigation of the electronic interaction between graphene and the C60 molecule it was found for the first time that the motion of the fullerene on graphene creates a small current that constantly changes due to the changing distance between the two objects. This physical phenomenon can be used as a physical principle for designing nanodevices.
 Please wait while we load your content...
Please wait while we load your content...

