DOI:
10.1039/C5RA11993F
(Paper)
RSC Adv., 2015,
5, 84578-84586
Influence of hydroxyapatite on thermoplastic foaming performance of water-plasticized poly(vinyl alcohol)
Received
22nd June 2015
, Accepted 28th September 2015
First published on 28th September 2015
Abstract
Poly(vinyl alcohol) (PVA)/hydroxyapatite (HA) composite foams suitable for bone tissue restoration were prepared through thermoplastic molding foaming using water as both a plasticizer and physical blowing agent. The effects of HA content on the foaming properties of the composites including the water states, water evaporation behaviors, interactions among the components as well as the cell structure and mechanical properties of the composite foams were studied. The results showed that HA nano-particles could form different hydrogen bonding or coordination interactions with water and PVA, thus making the bounded water in the system increase to a larger extent and the phase transition temperature of free water shift from 0.24 °C to −1.26 °C, consequently changing the relative content of different water. In this way, more nonfreezing water was formed, which was beneficial to the controllable foaming of water. HA particles could also act as a heterogeneous nucleating agent, thus effectively increasing the number of heterogeneous nucleating points in the system. With an increase in the HA content, foams with a more dense and uniform cell structure were obtained, and the mean size of the cell decreased from 760 μm (HA 0 wt%) to 270 μm (HA 20 wt%). The addition of HA improved the compression yield strength and compression modulus of the composite foams.
Introduction
With the development of society and economy, population aging has become a global social problem. Osteoarthritis caused by degeneration of joint cartilage is the most common health problem for an aging population,1–3 and the main treatment methods are joint replacement and restoration of the degradation parts. Among them, restoration of the degradation parts is relatively simple and can effectively reduce the patients’ pain at a low price. Accordingly, the development of new bone repair materials has become the focus of research at home and abroad.4,5
Poly(vinyl alcohol) (PVA) is a multi-hydroxyl polymer with excellent comprehensive properties, such as stable chemical properties, high mechanical strength, good compatibility with the human body and bio-ceramics, which can effectively solve the problem of low mechanical strength and the weak connection with bone tissue. Therefore, PVA has been considered to be an ideal material for cartilage replacement. Hydroxyapatite (HA), which contributes 65–70% to the bone matrix,6 is a bioactive ceramic and has good bioactivity and biocompatibility. In a physiological environment, HA can form chemical bonds with biological tissues through biological chemical reactions on its surface and induce the growth of human bone tissue. So, the PVA/HA composite is highly popular in the field of bone repair.7–11 However, ascribing to the quite close melting point (226 °C) and decomposition temperature (200–250 °C) of PVA,12 the present ways to prepare PVA/HA composites are almost based on aqueous solution. For example, Adolfo S. M.13 et al. manufactured HA reinforced PVA hydrogel by a freezing/thawing technique and found that the incorporation of HA decreased the degree of crystallinity, water absorption as well as the melting point and structural water content of the PVA hydrogel. The best mechanical properties, including the lowest friction coefficient and highest resistance strength were obtained for samples with 3% HA. Jimena S. Gonzalez14 et al. studied the effect of HA on the mechanical properties of a PVA hydrogel prepared by freezing–thawing and found that the addition of HA modified the physical and chemical features of the hydrogel and promoted crosslinking and the stability of the material, thus improving the mechanical properties of the hydrogels. However, the above reported aqueous methods to prepare PVA/HA composites were complex, and the strength and modulus, as well as the dimensional stability of the obtained composites, might still not meet the requirements of cartilage repair materials. So, novel methods to prepare high performance PVA/HA composites suitable for cartilage repair need to be developed.
In this paper, according to the structural characteristics of human bones, and based on the established thermal processing and foaming technologies of PVA in our research group,15–20 PVA/HA composite foams with the optimum pore size to promote cell adhesion, proliferation and colonization, i.e. 100–500 μm,6,21–23 were manufactured by using water as both a plasticizer and physical blowing agent through thermoplastic molding foaming. The effects of the HA content on the water states in the system, the interactions among the components, water evaporation behaviors, cell structure and mechanical properties were systematically studied. This is a big progress in the preparation of PVA/HA composite foam, which has potential applications in the field of bone tissue restoration.
Experimental section
Materials
Polyvinyl alcohol (PVA) (degree of polymerization is 1750 ± 50, hydrolysis degree is 99%) was purchased from Sichuan Vinylon Works, SINOPEC (China).
Hydroxyapatite (HA, biological reagent) was supplied by Nanjing Duly biological Co., LTD (China) with an average particle size of 20 nm.
Deionized water was used throughout the experiment.
Sample preparation
Quantified water and HA were mixed to form a stable suspension under the effect of ultrasonic oscillation. A certain amount of dried PVA was then added into the suspension, the mixture was uniformly blended to let the solutions completely swell into PVA at ambient temperature in a sealed vessel.
The swelled PVA/HA composites with 35 wt% water were first filled into an airtight rectangle metal frame (12 × 12 × 2 mm, self-made) and then heated at 170 °C for 15 minutes to melt the plasticized PVA/HA composites in a hot press machine. The molding pressure was 10 MPa. After that, the clamping force of the hot press was immediately released from 10 MPa to atmospheric pressure. The formed pressure difference made the composites foam. All the samples were prepared under the same conditions to avoid the extra impact factor to the morphology of the composite foam.
Characterization
Differential scanning calorimetry (DSC) test. The melting behaviors of water absorbed in the PVA/HA composite systems were analyzed by using a TA Q20 thermal analyzer (TA Instrument, Co., USA) at a heating rate of 5 °C min−1 from −40 °C to 15 °C under a nitrogen atmosphere with a flow rate of 50 ml min−1. The sample weight was about 6 mg.
Thermal gravity analysis (TG). Isothermal mass loss of the composite systems was studied by using a TA Q50 thermogravimetric analyzer (TA Instrument, Co., USA) under a nitrogen atmosphere with a flow rate of 50 ml min−1. The samples were first quickly heated from an ambient temperature to 150 °C at a heating rate of 40 °C min−1 and then fixed at 150 °C to collect the relationship between mass loss and time.
Fourier transform infrared spectroscopy (FT-IR) measurement. The interactions of the composite systems were tested on a Nicolet 6700 IR spectrometer (Thermo Scientific company, USA) in the ATR mode at room temperature. The tested samples were obtained by hot pressing the composite systems into films on a semi-automatic pressure molding machine at 130 °C. In order to keep the consistency of water content in all samples, the films were first dried in a vacuum drying oven at 80 °C until no weight loss, and then were supplied with 35 wt% moisture. The average thickness of the films was 80 μm.
Scanning electron microscopy (SEM) observation. The cell morphologies of the composite foams were observed on an Inspect (FEI, Japan) SEM Instrument at accelerating voltage of 5 kV. The foams were first freeze-fractured and then sputtered through a gold coating.
Expansion ratio measurement. The expansion ratio (ϕ) of the foam was evaluated as a specific ratio of the foam thickness and frame thickness (2 mm) using eqn (1) below:| |
 | (1) |
where ϕ is the expansion ratio, and hf and hm are the thicknesses of the expanded foam and metal frame (2 mm), respectively.
Cell density measurement. Cell density was assessed using eqn (2) as follows:24| |
 | (2) |
where n is the number of cells in the SEM micrograph, and A and M are the area and magnification factors of the micrograph, respectively.
Compression strength test. Compression properties of the composite foams were measured on a INSTRON4302 universal testing machine (Instron Corp., USA) equipped with a 30 kN load. The crosshead speed was 1 mm min−1. Each sample was tested five times.
Result and discussion
Water states
As is well known, the boiling point of bulk water is about 100 °C under atmospheric conditions. So it is quite difficult to obtain PVA foams with a satisfying cell structure when using water as a physical blowing agent due to its tempestuous evaporation at the foaming temperature of 170 °C. The key to achieve a good cell structure is the controllable evaporation of water, which is directly related to the water states in the system.
PVA is a multi-hydroxyl polymer, so water in PVA usually exists as three states, i.e. free water, freezable bound water and nonfreezing water, by forming different hydrogen bonding arrangements with the hydroxyl groups of PVA. Free water almost has no interactions with PVA, so presents similar thermodynamic behaviors, such as melting temperature, crystallization temperature and enthalpy, to bulk water, and can rapidly evaporate at 100 °C; freezable bound water has weak hydrogen bonding with PVA, thus showing a lower phase transition temperature and transition enthalpy than those of bulk water, and must absorb more heat energy to destroy these hydrogen bonds to transform into free water and then evaporate; while nonfreezing water has strong hydrogen bonding with PVA, therefore has no phase transition in the range of −100–0 °C (ref. 25) as well as a high evaporation temperature and low evaporation rate, and is quite difficult to be evaporated. Accordingly, the free water and freezable bound water in system usually take the role of the foaming agent, and the nonfreezing water mainly acts as a plasticizer. The water states in PVA can be influenced by many factors, such as the initial water content,26 added modifiers,27 etc. To avoid uncertainty and only investigate the influence of HA content on water states, all other parameters, like the initial water content, were set the same.
Fig. 1 shows the DSC curves of the PVA/water and PVA/water/HA composites, and the corresponding thermal data were listed in Table 1. In order to intuitively analyze the changes of each kind of water in the systems with varied HA content, the equations below were adopted to calculate the mass of free water (Wf), freezable bound water (Wfb) and nonfreezing water (Wnb), respectively.28
| | |
Wfb = (Ws × ΔHfb)/ΔH0
| (3) |
where Δ
Hfb and Δ
Hf respectively represent the phase transition enthalpy difference of the freezable bound water and free water in swelled PVA during the heating-up process; Δ
H0 is the phase transition enthalpy difference of pure water per unit mass,
i.e. 333.5 J g
−1;
28 Ww is the weight of the whole initial water and
Ws is the weight of the swelled PVA.
 |
| | Fig. 1 DSC curves of the PVA/water/HA composites (HA content: (a) 0%; (b) 1 wt%; (c) 5 wt%; (d) 10 wt%; (e) 15 wt%; (f) 20 wt%). | |
Table 1 Classification of the water states in the PVA/HA composites
| HA content/wt% |
Free water |
Freezable bound water |
Nonfreezing water |
| Peak 1 (°C) |
Enthalpy/J g−1 |
Vf/% |
Peak 2 (°C) |
Enthalpy/J g−1 |
Vfb/% |
Vnb/% |
| 0 |
0.24 |
6.00 |
5.14 |
−12.82 |
25.54 |
21.88 |
72.98 |
| 1 |
0.07 |
6.35 |
5.50 |
−13.88 |
24.38 |
21.10 |
73.40 |
| 5 |
−0.20 |
10.44 |
9.39 |
−14.08 |
16.95 |
15.24 |
75.37 |
| 10 |
−0.38 |
11.78 |
11.10 |
−14.21 |
8.043 |
7.58 |
81.32 |
| 15 |
−0.65 |
13.35 |
13.15 |
−14.24 |
3.493 |
3.44 |
82.99 |
| 20 |
−1.26 |
13.73 |
14.11 |
— |
— |
— |
85.89 |
The weight ratio of each water to whole initial water was calculated by the following equation:
where
i represents free water (f), freezable bound water (fb) or nonfreezing water (nf). The calculated data are respectively listed in
Table 1.
Obviously, two endothermic peaks appeared in the DSC curve of the PVA/water system (curve a) within the scope of −40 °C to 10 °C, which respectively represented the phase transition peaks of the free water (peak 1) and the freezable bound water (peak 2). With the addition of HA (Ca10(PO4)6(OH)2), the small volume and large specific surface area endowed the HA particles with a greater chance to interact with water molecules than the long chain PVA molecules, leading to the number of water molecules interacting with PVA in the PVA/water/HA system becoming less than that in the PVA/water system. The possibility of HA in forming interactions with water could be attributed to the OH and Ca2+ on its surface. However, these interactions might be weaker than those between PVA–water, which would be verified in later FTIR testing, thus making the water that only interacted with HA more like bulk water, which was reflected in the DSC curve by the phase transition peak of free water in the system moving to a lower temperature and the peak area increasing, i.e. more free water formed in the system. Correspondingly, the residual water got more chance to directly form strong hydrogen bonding with PVA, leading to the increase of the nonfreezing water content in the system. Moreover, the existent interaction model of PVA–water–HA could further restrict water molecules, also contributing to the increase in the content of nonfreezing water.
With an increase in the HA content, more and more water interacted with HA, making the phase transition temperature of free water shift more to a lower temperature and its content further increased. Contrarily, the water interacting with PVA further decreased and the hydrogen bonding between them enhanced, resulting in the increase of the content of nonfreezing water. The changes of the interactions among each component in the system led to the reduction of the content of the freezable bound water, which was reflected in the DSC curves by its phase transition peak area decreasing with an increase in HA content, and disappearing when the HA content reached 20 wt%.
Intermolecular interactions
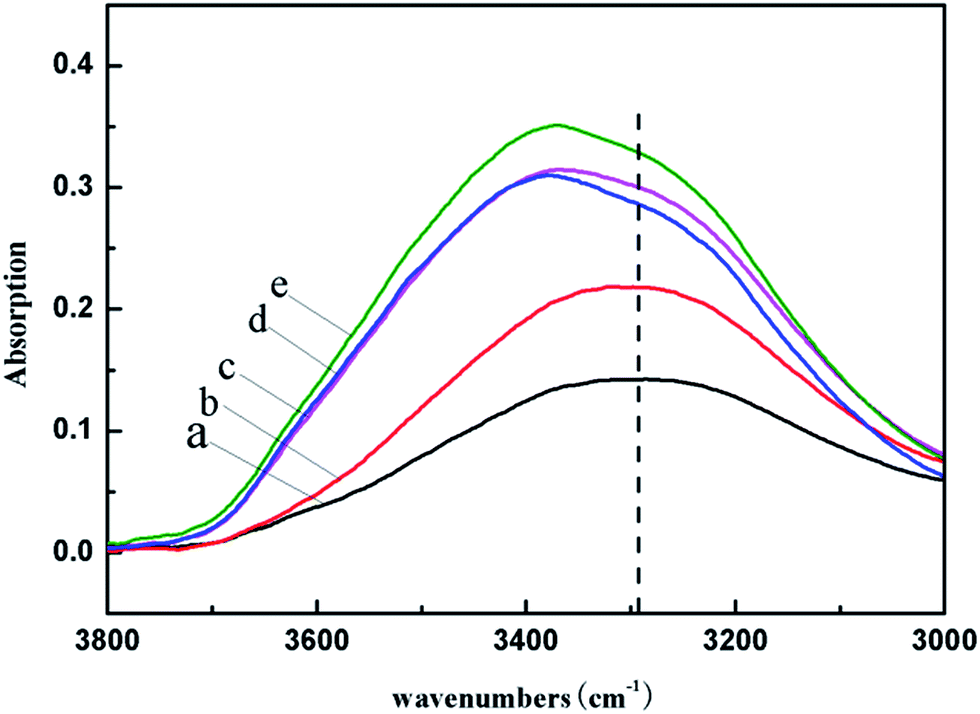
The constraint of HA to the water in system came from the hydrogen bonding or the coordination interactions among the HA, PVA and water molecules. To further illustrate the influence of HA on the interactions in the systems, the FT-IR spectra of pure PVA, HA, PVA/HA, PVA/water and PVA/water/HA were analyzed, as shown in Fig. 2. It could be seen from Fig. 2 that pure PVA had a fairly wide hydroxyl stretching band in the range of 3000–3800 cm−1, attributed to the free alcohol (unbounded –OH stretching band at ν = 3600–3650 cm−1) in the amorphous phase and the bonded hydroxyl (ν = 3200–3570 cm−1) in the crystalline phase.29,30 Compared with pure PVA, the hydroxyl stretching band of PVA/water (curve a) shifted to a higher wavenumber, indicating that the addition of water weakened the original inter- or intra-molecular hydrogen bonding among the hydroxyl groups of PVA and the newly formed hydrogen bonding between PVA and water was weaker. When incorporating HA into the PVA/water system, the relatively weak interactions between HA and water made the hydroxyl stretching band of the system shift further to higher wavenumbers, and the greater the HA content, the greater the deviation of the hydroxyl stretching band. Simultaneously, the hydroxyl stretching band became wider with the increase in HA content, which was ascribed to the increase of the types of hydrogen bonding, which can be seen in Fig. 3.
 |
| | Fig. 2 FT-IR spectrum of PVA, HA, PVA/HA and PVA/water/HA composites with different HA content: (a) 0%; (b) 1 wt%; (c) 5 wt%; (d) 10 wt%; (e) 20 wt%. | |
 |
| | Fig. 3 Scheme of hydrogen bonding in PVA/water (A) and PVA/water/HA (B). | |
The wide peaks in the range of 3000–3800 cm−1 consisted of the hydroxyl stretching bands of PVA and/or HA and/or water. To observe these peaks more clearly, we enlarged them, as shown in Fig. 4. Obviously, the PVA/water system showed a steamed bread shaped hydroxyl stretching band (curve a), which could be attributed to the continuous superposition of hydrogen bonding with different strength between water and PVA.16 With the addition of HA, this peak became wider, and gradually presented a shoulder peak at about 3290 cm−1. To intuitively see the changes of each peak, we deconvolved the peaks of curve a, d and e in Fig. 4 by using the professional software Peakfit v4.12. The fitting degree reached 0.999 and the standard error was less than 10−3. The fitting curves are shown in Fig. 5, and the relative content of the three states of water and the peak position are listed in Table 2. According to ref. 31, the peaks centered at 3186 cm−1, 3498 cm−1 and 3599 cm−1 were the hydroxyl stretching bands of nonfreezing water, freezable bound water and free water, respectively. Apparently, with the increase in HA content, both the strength of the hydroxyl stretching bands of the nonfreezing water and free water in the system increased, indicating an increase in the content of nonfreezing water and free water, while for freezable bound water, the strength of its hydroxyl stretching band gradually dropped and the peak disappeared when the HA content reached 20 wt%, meaning that only free water and nonfreezing water existed in the system. The results of the deconvolution of the FT-IR spectra, cross referenced with the DSC analysis, indicate that the relative content of the three states of water in the system could be adjusted through the special interactions among water, HA and PVA, which is beneficial in controlling the water content used for foaming and plasticization, as well as the cell structure of the final foams.
 |
| | Fig. 4 FT-IR spectra of the PVA/water/HA composites from 3000–3800 cm−1 (HA content: (a) 0%; (b) 1 wt%; (c) 5 wt%; (d) 10 wt%; (e) 20 wt%). | |
 |
| | Fig. 5 Simulated FT-IR spectra of the PVA/water/HA composites with 0 wt% HA (A), 10 wt% HA (B) and 20 wt% HA (C). | |
Table 2 The weight percentage of the different kinds of water in the PVA/water/HA composites obtained from the simulated FT-IR spectra
| HA content/wt% |
Nonfreezing water |
Freezable bound water |
Free water |
| Weight percentage/wt% |
Wavenumber/cm−1 |
Weight percentage/wt% |
Wavenumber/cm−1 |
Weight percentage/wt% |
Wavenumber/cm−1 |
| 0 |
73.3 |
3186.1 |
20.1 |
3498.0 |
6.7 |
3598.8 |
| 10 |
80.6 |
3190.0 |
8.4 |
3496.9 |
11.1 |
3598.2 |
| 20 |
84.6 |
3191.6 |
— |
— |
15.4 |
3597.5 |
Evaporation behaviors of water
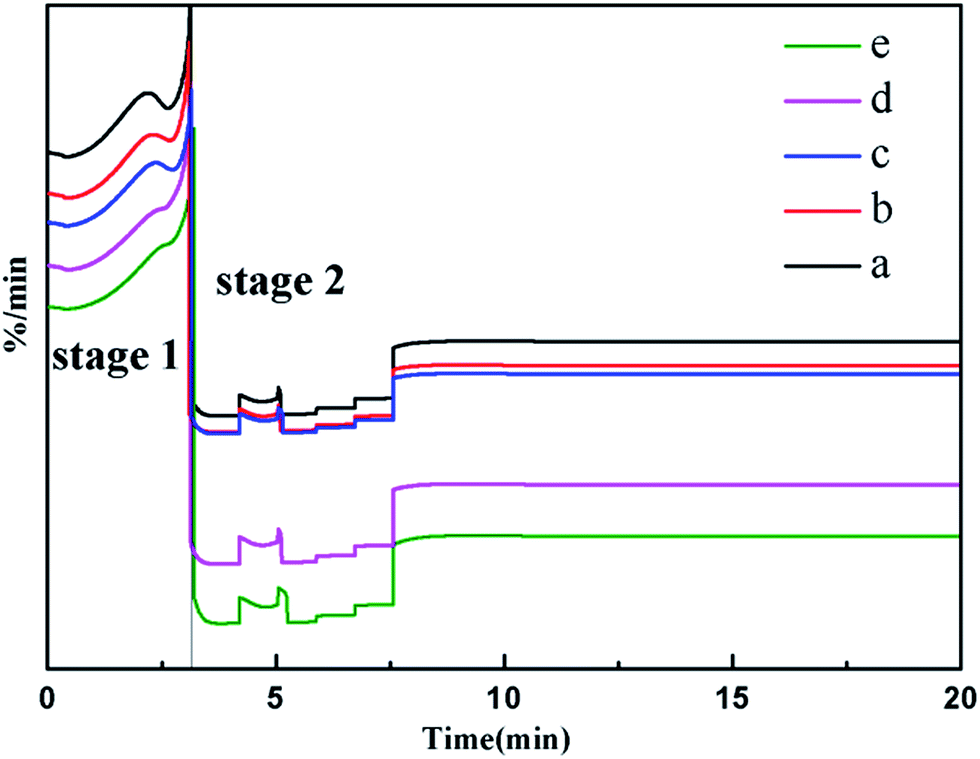
The changes of the water states in the system with the addition of HA certainly influenced the evaporation of water, which could be directly seen from the TG testing. Fig. 6 shows the isothermal weight loss behaviors of PVA/water and PVA/water/HA. Considering the high decomposition temperature of PVA and HA (>200 °C), the weight loss in Fig. 6 could be ascribed to water evaporation rather than the decomposition of PVA or HA at the testing temperature, i.e. 150 °C. In order to more intuitively analyze the evaporation of water, the mass of PVA and HA were reduced and the corresponding derivative thermogravimetry curves of water at different times were obtained, as shown in Fig. 7. Apparently, based on the evaporation rate in Fig. 7, the evaporation curve of water in each system could be divided into two stages: rapid evaporation (stage 1) and slow evaporation (stage 2). Free water with no interactions with PVA or freezable bound water with weak interactions with PVA mainly evaporated at the first stage due to their low evaporation energy barrier. With extension of the heating time, more and more energy was absorbed by the system, leading to the evaporation of nonfreezing water starting. However, ascribing to the strong hydrogen bonding of the nonfreezing water with PVA, this part of the water was relatively difficult to be evaporated and had a low evaporation rate, making the weight loss ratio of the system gradually tend to a balance, as seen in stage 2 of the figures. After the addition of HA, water evaporation was further inhibited due to the formation of more hydrogen bonding and the coordination interactions among water, HA and PVA, which is reflected in the TGA curves as with the increase in HA content, both the water evaporation rate and the evaporation capacity reduced. Accordingly, the addition of HA was beneficial in adjusting the water states and their evaporation behaviors, thus realizing the controllable foaming of water.
 |
| | Fig. 6 Evaporated water content vs. initial water content in the PVA/water/HA composites at 150 °C (HA content: (a) 0%; (b) 1 wt%; (c) 5 wt%; (d) 10 wt%; (e) 20 wt%). | |
 |
| | Fig. 7 DTG curves of the PVA/water/HA composites at 150 °C (HA content: (a) 0%; (b) 1 wt%; (c) 5 wt%; (d) 10 wt%; (e) 20 wt%). | |
Cell morphologies
Normally, the formation of foam includes three stages: initial nucleation, bubble growth and stabilization, and solidification of the resin matrix.32 During the nucleation, for the PVA/water system, a uniform melt was obtained after being heated in the mold because of the absence of a nucleating agent, so the cell was mainly formed through homogeneous nucleation after the pressure relief, resulting in the low cell number and uneven cell size, as shown in Fig. 8a. With the addition of HA nano-particles, their heterogeneous nucleation effect and good dispersity in the PVA system33 surely created more nucleation sites and made more bubbles nucleate on their surface during the process of pressure relief, leading to a smaller cell size (Fig. 8b–e and 9b), a higher cell density (Fig. 9c) and a lower expansion ratio (Fig. 9a). Furthermore, the relatively higher melt viscosity of the PVA/water/HA composite system compared to the PVA/water system33 was helpful in preventing the collapse of bubbles during the foaming process, as well as the resin solidification afterwards. With the increase in HA content, more and more nucleation sites formed in the system, and as a result, the cell number increased, the cell size decreased and the foam became more and more uniform. For example, the mean cell diameter of the composite foam with 20 wt% HA was 270 μm, which was much smaller than that of the foam without HA, i.e. 760 μm.
 |
| | Fig. 8 SEM photos of the PVA/HA composite foams (HA content: (a) 0%; (b) 1 wt%; (c) 5 wt%; (d) 10 wt%; (e) 20 wt%). | |
 |
| | Fig. 9 Expansion ratio (a), cell size (b) and cell density (c) of the PVA/HA composite foams. | |
For the PVA/water system, when heated at 170 °C, water in the system with no interaction with PVA (free water) or with a weak interaction with PVA (freezable bound water) could absorb enough energy to escape the PVA and transform into gas so as to provide the pressure relief for foaming; while water with strong hydrogen bonding with PVA (nonfreezing water) needed more heat energy to evaporate, so this part of the water could act as a plasticizer in the system in certain times to adjust the viscosity and the melt strength of the matrix, as well as its penetration ability for water vapor. The addition of HA enhanced the nonfreezing water in the system by forming hydrogen bonds and coordination with water and the OH groups of PVA through its –OH and Ca2+, thus increasing the content of plasticizer for PVA and decreasing the content of blowing agent at the same processing conditions. The blowing agent and the plasticizer in the system mutually transformed during the foaming process, adjusting the melt viscosity and strength as well as the content of blowing agent, thus deciding the foaming properties of the matrix. In other words, a foam material with a certain cell size could be obtained by adjusting the HA content to control the water state in the system and the melt viscosity of the system.
Compression properties
Having appropriate compression properties is a basic requirement of a tissue engineering scaffold. Fig. 10 shows the compression tests of PVA/HA composites, the compression modulus was calculated as the slope of 0% to 10% deformation of the compression. Apparently, both the compression strength and compression modulus of the PVA/HA composite foams increased with the increase in HA content and reached a climax when the HA content was 10 wt%. For example, the compression modulus and compression yield strength respectively increased from 4.52 ± 0.47 MPa and 1.01 ± 0.09 MPa for the foam without HA to 7.83 ± 0.32 MPa and 2.28 ± 0.07 MPa for the composite foam with 10 wt% HA. Two reasons might be attributed to the compression properties of the composite foams: the cell structure and the dispersion of HA in the matrix. The composite foam with 10 wt% HA was appropriate to get both the proper number of nucleation sites and the best dispersion effect, thus achieving a uniform cell structure and high cell density as well as the best compression properties.
 |
| | Fig. 10 Compression strength (A), compression modulus and compression yield (B) of the PVA/HA composite foams. | |
Conclusions
PVA/HA composite foams were first prepared through thermoplastic processing by using water as a physical blowing agent and plasticizer. The –OH and Ca2+ on the surface of the nano-HA particles could form strong hydrogen bonds and coordination interactions with water and PVA molecules, thus manipulating the states of water in the system and altering the relative contents of the three kinds of water. With the addition of nano-HA particles, the total content of free water and freezable bound water used as the physical blowing agent decreased; meanwhile, the content of nonfreezing water, working as a plasticizer, increased. The manipulation of the water states and their content improved the thermoprocessability of PVA and made water evaporation controllable for foaming. Moreover, nano-HA particles worked as a heterogeneous nucleating agent in the system, thus transferring the system from homogeneous nucleation to heterogeneous nucleation and lowering the surface free energy barrier of the composite system for foaming. In this way, composite foams with a cell size of 100–500 μm, a uniform structure and high compression properties were achieved, suggesting great promise for bone tissue-engineering applications.
Acknowledgements
The authors greatly acknowledge the financial support of the International Science & Technology Cooperation Program of China (2013DFG52300) and the National Natural Science Foundation of China (51121001).
References
- K. Burg, S. Porter and J. F. Kellam, Biomaterials, 2000, 21, 2347–2359 CrossRef CAS.
- R. J. Covert, R. D. Ott and D. N. Ku, Wear, 2003, 255, 1064–1068 CrossRef CAS.
- S. Miyata, K. S. Furukawa, T. Ushida, Y. Nitta and T. Tateishi, Mater. Sci. Eng., C, 2004, 24, 425–429 CrossRef PubMed.
- G. Georgiou, L. Mathieu, D. P. Pioletti, P. E. Bourban, J. A. E. Manson, J. C. Knowles and S. N. Nazhat, J. Biomed. Mater. Res., Part B, 2007, 80, 322–331 CrossRef CAS PubMed.
- L. J. White, V. Hutter, H. Tai, S. M. Howdle and K. M. Shakesheff, Acta Biomater., 2012, 8, 61–71 CrossRef CAS PubMed.
- A. J. Salgado, O. P. Coutinho and R. L. Reis, Macromol. Biosci., 2004, 4, 743–765 CrossRef CAS PubMed.
- Y. Pan and D. Xiong, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2010, 25, 474–478 CrossRef CAS.
- Y. Pan and D. Xiong, Mech. Time-Depend. Mater., 2013, 17, 195–204 CrossRef CAS.
- D. Qu, J. Li, Y. Li, A. Khadka, Y. Zuo, H. Wang, Y. Liu and L. Cheng, J. Biomed. Mater. Res., Part B, 2011, 96, 9–15 CrossRef PubMed.
- N. S. Ibrahim, G. Krishnamurithy, H. R. B. Raghavendran, S. Puvaneswary, W. M. Ng and T. Kamarul, Mater. Lett., 2013, 113, 25–29 CrossRef CAS PubMed.
- Y. Guo, J. Guo, D. Bai, H. Wang, X. Zheng, W. Guo and W. Tian, Artif. Organs, 2014, 38, 580–586 CrossRef CAS PubMed.
- T. Tanigami, H. Hanatani, K. Yamaura and S. Matsuzawa, Eur. Polym. J., 1999, 35, 1165–1171 CrossRef CAS.
- A. Sebastian Maiolo, M. Nicolas Amado, J. Soledad Gonzalez and V. Alejandra Alvarez, Mater. Sci. Eng., C, 2012, 32, 1490–1495 CrossRef PubMed.
- J. S. Gonzalez and V. A. Alvarez, J. Mech. Behav. Biomed. Mater., 2014, 34, 47–56 CrossRef CAS PubMed.
- R. Wang, Q. Wang, L. Li and Z. K. Hua, Polym. Mater.: Sci. Eng., 2001, 111–113 CAS.
- R. Wang, Q. Wang and L. Li, Polym. Int., 2003, 52, 1820–1826 CrossRef CAS PubMed.
- N. Chen, L. Li and Q. Wang, Plast., Rubber Compos., 2007, 36, 283–290 CrossRef CAS PubMed.
- L. Li, Q. Wang and R. Wang, J. Appl. Polym. Sci., 2005, 98, 774–779 CrossRef CAS PubMed.
- X. H. Xiong, F. Yang, Q. Wang and L. Li, China Plast. Ind., 2007, 10–12 CAS.
- Q. Wang, L. Li, N. Chen, Y. Liu and S. B. Bai, Acta Polym., 2011, 932–938 CrossRef.
- C. E. Petrie Aronin, K. W. Sadik, A. L. Lay, D. B. Rion, S. S. Tholpady, R. C. Ogle and E. A. Botchwey, J. Biomed. Mater. Res., Part A, 2009, 89, 632–641 CrossRef PubMed.
- K. F. Leong, C. K. Chua, N. Sudarmadji and W. Y. Yeong, J. Mech. Behav. Biomed. Mater., 2008, 1, 140–152 CrossRef CAS PubMed.
- V. Karageorgiou and D. Kaplan, Biomaterials, 2005, 26, 5474–5491 CrossRef CAS PubMed.
- N. Zhao, L. H. Mark, C. Zhu, C. B. Park, Q. Li, R. Glenn and T. R. Thompson, Ind. Eng. Chem. Res., 2014, 53, 11962–11972 CrossRef CAS.
- L. Daniliuc and C. David, Polymer, 1996, 37, 5219–5227 CrossRef CAS.
- Y. L. Li, L. Li, N. Chen and Q. Wang, Suliao, 2011, 1–4 Search PubMed.
- X. Wang, L. Li, N. Chen and Q. Wang, Chemical Journal of Chinese Universities, 2012, 813–817 Search PubMed.
- C. Rongshi, W. Li and X. Feng, Polymer, 2005, 46, 12026–12031 CrossRef PubMed.
- H. S. Mansur, R. L. Orefice and A. Mansur, Polymer, 2004, 45, 7193–7202 CrossRef CAS PubMed.
- L. Daniliuc, C. Dekesel and C. David, Eur. Polym. J., 1992, 28, 1365–1371 CrossRef CAS.
- Z. H. Ping, Q. T. Nguyen, S. M. Chen, J. Q. Zhou and Y. D. Ding, Polymer, 2001, 42, 8461–8467 CrossRef CAS.
- D. Eaves, Handbook of polymer foams, Shawbury UK, 2004 Search PubMed.
- X. Chen, N. Chen and Q. Wang, Polym. Bull., 2013, 26–32 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.