
Interlocked dimerization of C3-Symmetrical boron difluoride complex: designing non-cooperative supramolecular materials for luminescent thin films†
Abstract
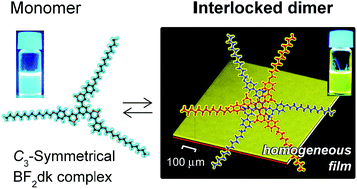
A lipophilic complex with radially-connected three β-diketonate boron difluoride (BF2dk) units to a central benzene ring was newly developed. The C3-symmetrical BF2dk complex (1) formed a self-complementary interlocked dimer (1)2 with increasing concentration in CHCl3 as revealed by NMR spectroscopy and quantum chemical calculations. A remarkable luminescence color change from blue to yellow was observed in response to the formation of interlocked dimers. Electrostatic interactions, hydrogen bonding between negative convexes (BF2 moiety) and positive concaves (three protons aligned on each arm) principally contribute to the dimerization, whereas the formation of interlocked dimers was accompanied by conformational changes of constituent molecules which interrupted further association. Consequently, casting of the chloroform solution of interlocked dimers on solid supports gave uniform thin films without uneven crystallization. It provides a new perspective for designing anti-cooperative systems for homogeneous molecular coatings.
 Please wait while we load your content...
Please wait while we load your content...

