
Ribbon-like and spontaneously folded structures of tungsten oxide nanofibers fabricated via electrospinning
Andrei Stanishevsky*a,
Joshua Wetuskia,
Michael Walocka,
Inessa Stanishevskayaa,
Hélène Yockell-Lelièvreb,
Eva Košťákovác and
David Lukášc
aDepartment of Physics, University of Alabama at Birmingham, 1300 University Boulevard, Birmingham, AL 35294, USA. E-mail: astan@uab.edu
bDépartement de Chimie, Université de Montréal, CP 6128 Succ. Centre-Ville, Montreal, Canada
cFaculty of Textile Engineering, Technical University of Liberec, Studentska 2, Liberec 1, 461 17, Czech Republic
First published on 10th August 2015
Abstract
Tungsten oxide (WO3) nanofibers with shapes ranging from cylindrical to ribbon-like were prepared by annealing electrospun polyvinylpyrrolidone/ammonium metatungstate (PVP/AMT) fibers. Formation of periodically folded “zigzag” patterns in PVP/AMT and WO3 ribbon-like fibers was observed for the first time at the initial stage of the electrospinning process on the surface of a stationary substrate. Among methods tested (capillary needle, needleless dc- and ac-electrospinning), only the capillary needle dc-electrospinning process was effective in producing ribbon-like fiber structures. Annealing of such PVP/AMT fibers at 500 °C in air led to the formation of 80 ± 10 nm thick WO3 ribbons with a width-to-thickness ratio of up to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Scanning Electron Microscopy (SEM), Transmission Electron Microscopy (TEM), X-ray diffraction (XRD), X-ray Photoelectron Spectroscopy (XPS), and Raman spectroscopy were used to analyze the material. Analyses revealed that regardless the fiber's shape, the annealed oxide fibers were polycrystalline with a grain size of 60 ± 30 nm and consisted of the monoclinic phase of WO3. When compared to cylindrical fibers, the ribbon-like WO3 nanofibers exhibited higher porosity but lower mechanical strength with increased width of the ribbon-like structure.
:1. Scanning Electron Microscopy (SEM), Transmission Electron Microscopy (TEM), X-ray diffraction (XRD), X-ray Photoelectron Spectroscopy (XPS), and Raman spectroscopy were used to analyze the material. Analyses revealed that regardless the fiber's shape, the annealed oxide fibers were polycrystalline with a grain size of 60 ± 30 nm and consisted of the monoclinic phase of WO3. When compared to cylindrical fibers, the ribbon-like WO3 nanofibers exhibited higher porosity but lower mechanical strength with increased width of the ribbon-like structure.
Introduction
In the last decade, electrospinning has been increasingly employed for the manufacture of a large variety of polymeric and inorganic fibrous materials with micro-to-nanoscale fiber diameters.1–3 Nearly every electrospinning process involves a polymeric precursor, and inorganic fibers are mostly obtained by thermal decomposition of the polymeric component in the fibers containing inorganic salts or other suitable compounds. Oxide nanofibers make up the largest group among electrospun inorganic nanofibrous materials. Many simple and complex oxide nanofibrous materials have been engineered as environmental sensors, enhanced catalysts and catalyst supports, anode materials for lithium batteries, and for other applications.2,4–6The application-specific performance of oxide nanofibrous materials depends on the surface microstructure, crystallinity, chemical and phase composition, shape, surface morphology, porosity, and interconnectivity of individual fibers. Generally, the shape of oxide nanofibers is determined by the shape of the original oxide precursor-loaded polymeric fiber, as well as by the ratio of the oxide precursor and polymer. The cylindrical shapes of electrospun polymeric fibers are common,7 but other fiber shapes, such as belts and ribbons have also been reported.8–10 Inorganic nanofibers with flat belt- and ribbon-like shapes have been made of several simple and complex oxides, for example, In2O3/Co3O4, Al2O3, BaTiO3, LaFeO3, RuO, and Bi2WO6.11–16 A larger surface-to-volume ratio of the belt- and ribbon-like nanofibers makes such fiber architectures attractive for gas sensors and catalyst supports. Furthermore, the belt- and ribbon-like geometries can lead to the changes in the fiber's pore shape and size distribution, introduce the anisotropy of transport and mechanical properties, and can subsequently result in the nanofibrous materials with better gas selectivity, multiferroic and optical properties.17–19 Currently, there are few reports that directly compare the properties of cylindrical nanofibers to other nanofiber shapes. For example, the enhancement of photoluminescence of nanobelt-like fibers with respect to cylindrical ones have been observed for Gd1.9Eu0.1MoO6 and HfO2.20,21
This paper presents results on the fabrication of polycrystalline WO3 ribbon-like nanofibers by thermal processing of the ribbon-like fibers prepared by electrospinning polyvinylpyrrolidone (PVP) loaded with ammonium metatungstate (AMT). Previously, WO3 nanofibers with different crystallinities and surface morphologies have been fabricated by electrospinning,22–26 but non-cylindrical fiber shapes have not been observed. WO3 nanofibers have demonstrated attractive characteristics in gas sensing,27–31 as well as photocatalytic,32 and electrochromic33 applications, and tailoring the WO3 nanofiber shape from cylindrical to a ribbon-like one could further their potential for such purposes. The present experiments showed that it is possible to prepare predominantly ribbon-like PVP/AMT and subsequent WO3 nanofibers by using a capillary needle dc-electrospinning method. Spontaneous formation of folded “zigzag” ribbon-like fiber patterns at the initial stage of the electrospinning process was observed, a phenomenon which can affect the electronic properties of WO3 in such structures and offer further insight into electrospinning phenomena and directed fabrication of various fiber microarchitectures.
Experimental details
The typical base precursor solution was made by mixing 5.0 mL 30% w/v solution of polyvinylpyrrolidone (PVP, Mw 1300000, Sigma-Aldrich) with 5.0 mL 30% w/v solution of ammonium metatungstate hydrate (AMT, Sigma-Aldrich) in ultrapure DI water. The resulting 15% w/v PVP/AMT solution with 1:1 PVP to AMT w/w ratio was stirred, sonicated, and aged for 24 hours. The solution's dynamic viscosity, surface tension, and electrical conductivity were determined using Wells–Brookfield LVDV-II + PRO cone/plate viscometer, Fisher Scientific capillary tensiometer and Mettler Toledo FEP30 conductivity meter, respectively. All measurements were done at 25 °C ambient temperature.
The capillary needle dc-electrospinning system included a syringe pump with a 5 mL plastic syringe directly connected to a 21 gauge capillary needle. The positive electrode of a 40 kV dc-power supply (Gamma High Voltage Research, Inc.) was attached to the needle. The substrates (p-type 10 ohm cm Si wafers, aluminum foil, glass, and paper) were placed on a grounded stainless steel collector located at the distance from 100 mm to 300 mm from the needle. The process was conducted either with or without a 300 × 300 × 300 mm vented plastic enclosure. The fibers were generated at the flow rate in the range of 0.1–0.25 mL h−1, and the dc-voltage varied from 12 kV to 30 kV.
Needleless dc-electrospinning was utilized with a vertical 10 mm diameter metal rod connected to the positive terminal of a dc-power supply. A layer of the PVP/AMT solution was formed on the top surface of the rod with a pipette, and the process was run for a few seconds to reduce the magnitude of change in the precursor viscosity due to solvent evaporation. The voltage and distance were similar to those in the capillary needle dc-electrospinning process.
Needleless ac-electrospinning utilized a simple setup with the vertical 10 mm diameter hollow metal rod. The polymer solution was supplied to the top surface of the rod through an infusion syringe pump. The ac-voltage was provided by a commercial step-up voltage connected to an autotransformer operated at 50 Hz/230 V. Further details of this method can be found in ref. 34 The PVP/AMT fibers were generated at the 25–30 kV rms ac-voltage. There was no grounded collector in this experiment. It has been suggested that a dynamic virtual counter electrode forms in the expanding flow of fibers due to the alternating nature of the electric field. The stationary insulated substrate was introduced into the flow of generated fibers at distances from 200 mm to 400 mm and withdrawn from the flow after a sufficient amount of fibers was accumulated.
Finally, a laboratory “Nanospider” apparatus by Elmarco was used to generate the fibers from the side surface of a rotating metal cylinder partially immersed in the PVP/AMT solution at 50 kV to 60 kV dc-voltage and the distance between 300–400 mm.35
The fabricated PVP/AMT fibers were annealed by heating the specimens in the air at a rate of 3 °C min−1 and holding them at 500 °C for 3 hours using a programmable furnace (Isotemp from Fisher Scientific).
The size, shape, and surface morphology of the fibers were investigated with field-emission scanning electron microscopy (FE-SEM, FEI Quanta 650). SEM imaging was done in secondary electron mode, with an accelerating voltage of 15 kV, electron probe current 2 μA, and a chamber pressure of 1 × 10−4 Pa. Chemical composition of the fibers was determined by the electron dispersive spectroscopy (EDS) accessory of SEM. During EDS, the accelerating voltage was 15 kV with an integration time of 300 s. Artistic representations were created by processing the SEM images of the folded zigzag patterns of ribbon-like fibers using Autodesk Maya 2013 (for structure modeling) and Adobe After Effects CC 2014 (for composing).
Transmission Electron Microscopy (TEM) imaging was performed using a JEOL JEM-2100F microscope under an acceleration voltage of 200 kV. The TEM samples of WO3 fibers were prepared on a formvar-coated nickel grid from an aqueous dispersion of the collected fibers.
Crystalline phase composition was determined using a Philips X'pert MPD thin-film X-ray diffractometer with a Cu Kα tube (wavelength 0.15406 nm) operating at 45 kV and 40 mA. The detector was scanned between 20° and 70°, with a constant take off angle of 5°. The experimental diffraction patterns were compared with the ICDD powder diffraction file (PDF) database.36
Chemical bonding in the samples was determined by a PHI 5000 Versaprobe imaging X-ray photoelectron spectrometer operating a monochromatic, focused Al Kα X-ray source (E = 1486.6 eV) at 25 W with a 100 μm spot size. All samples were grounded. Charge neutralization was provided by a cold cathode electron flood gun and low-energy Ar-ions. All measurements were taken at room temperature and at a pressure of 2 × 10−6 Pa (the system base pressure was 5 × 10−8 Pa). Surface sputtering was not generally used in these experiments to avoid a possibility of the change in the chemical bonding state associated with ion-induced damage. Multipak v9.0 software was used to analyze the resulting spectra. To verify the presence of residual carbon only (detection limit 0.1 at%), Ar-ion sputter etching was used for a period of 1 min at 1 kV accelerating voltage. This procedure removed about a 3.0 nm layer from the surface.
FTIR spectra (Vertex 70 spectrometer, Bruker Optics) were acquired in transmission mode directly from PVP/AMT and WO3 nanofiber samples on intrinsic silicon substrate, and the substrate background spectra were subtracted. In all cases, the FTIR spectra represented an average of 100 scans recorded with a resolution of 4 cm−1 for each sample.
Raman spectra were collected from several locations across each sample using Dilor XY microRaman spectroscopy system equipped with a 300 mW solid state laser (Dragon Lasers), operating at λ = 532 nm and focused to a 10 μm spot size. All measurements were taken in standard laboratory conditions.
Results and discussions
In a typical electrospinning experiment conducted with an aqueous precursor solution of polyvinylpyrrolidone (PVP, Mw ∼ 1.3 M) and ammonium metatungstate (AMT) mixed in various ratios and concentrations, thin mats of randomly oriented intertwined cylindrical fibers with diameters from 200 nm to 10 μm were produced, depending on the electrospinning method and process parameters. This result was consistent with other reports.22–26 In present experiments, ribbon-like PVP/AMT fibers have been observed for the first time as a dominant fiber shape in a capillary needle dc-electrospinning method from an arbitrarily high concentration (15 w% PVP) of the precursor solution containing the polymer and tungsten salt in a 1:1 w/w ratio (Fig. 1a). This precursor was characterized by 1270 ± 30 cP magnitude of viscosity, 64.5 ± 2.5 mN m−1 surface tension, and 10.2 ± 0.01 mS cm−1 electrical conductivity. The maximum yield of ribbon-shaped fibers was achieved only within a narrow range of the needle dc-electrospinning process parameters at this precursor concentration. The needle-collector distance in the range of 200 mm and 250 mm, and voltage between 20 kV and 22 kV, played the critical role in achieving the largest yield of ribbon-like structures. However, so far there has been little to no success in obtaining ribbon-like PVP/AMT fibers with needleless methods.34,35,37 Only accidental clumps of ribbon-like fibers (Fig. 1c) were observed when dc-electrospun from a thin layer of precursor put on the top surface of a vertical 10 mm diameter metal rod. This could be an effect of the solvent evaporating from the precursor layer and its increasing concentration and viscosity during the process. Interestingly, the cylindrical fibers (1–3 μm diameter) in this case formed flat cable-like assemblies. Significantly higher ac-voltage (>30 kV rms) was needed to electrospin the PVP/AMT with a similar alternating current electrospinning setup.34 The ac-electrospun PVP/AMT fiber diameter was significantly larger (up to 10 μm) than in other experiments, and only few ribbon-like fibers with the width of up to 15–20 μm were observed (Fig. 1d). Only cylindrical, partially fused fibers were obtained when the PVP/AMT precursor was dc-electrospun from the side surface of the rotating cylinder in a free-surface “Nanospider” process, which used a much higher voltage (Fig. 1b).35 These results show that the initial viscosity, surface tension, and electrical conductivity of the precursor and process parameters are important in tailoring the shape of fibers. However, the alteration of rheological properties in the forming jet and the electric field distribution near the electrode surface (restrained by the needle or free-surface electrode geometries) can also be the critical factors for the formation of ribbon-like structure of the fibers.
 | ||
| Fig. 1 Representative SEM images of PVP/AMT fibers prepared from a 15% PVP/AMT 1:1 precursor solution by (a) capillary needle, (b) “Nanospider” free-surface dc-electrospinning, (c) needleless dc- and (d) needleless ac-electrospinning methods. | ||
Thus, further investigation focused on capillary needle dc-electrospinning as the preferred method to prepare ribbon-like PVP/AMT fibers and their analysis. For the PVP/AMT precursor and the process conditions used in the experiments, the ribbon-like fiber thickness was always approximately 150 nm and rather uniform, whereas the width of the ribbons varied from 1.1 μm to 4.0 μm. Some typical cylindrically-shaped fibers with diameters of 650 ± 200 nm were also still present (Fig. 1a).
The formation of ribbon-like fibers during the electrospinning process has been observed and modeled for a number of polymer systems.8–10 The basic processes leading to flat fiber structures involve rapid solvent evaporation and formation of a thin solidified surface layer (“skin”) in the propagating polymer jet, followed by evaporation of the residual solvent and then collapse of the resulting hollow tubular shape due to a combination of atmospheric pressure, cohesive forces, charge redistribution and material stiffness. Based on the data of Reneker and Yarin,9 the uniformity and rather sharp edges of the fabricated PVP/AMT ribbon-like fibers can indicate relatively low stiffness of this material. Furthermore, the width of the ribbon, which is roughly half of the “skin” length, allows the estimation of jet diameter when the “skin” is formed. In present experiments, this gave the jet diameter of 1.7 ± 1.0 μm at that instant. The residual solvent seemed to gradually evaporate through the solidified PVP/AMT layer without rupturing the tubular shape as it was observed by Su et al.21 for HfO2 nanobelts. This led to the continuous and uniform ribbon-like structure of the resulting fibers. The cross-sectional area of the ribbon-like PVP/AMT fibers varied from 0.17 μm2 to 0.6 μm2, the same as the solid cylindrical fibers with diameters in the range of 460 nm to 870 nm would have. The latter numbers are close to the actual diameters of cylindrical fibers that were occasionally present in the ribbon-like fibrous material.
Although the electrospun PVP/AMT ribbon-like fibers usually formed a random fibrous layer, they were also found to arrange into folded zigzag patterns along with other less organized structures on the exposed surface of the collector as shown in Fig. 2a and b. This happens when the fibers collapse into a periodic pattern as they approach and are stopped by the surface. The folded zigzag patterns were only observed for the ribbon-like fibers, and cylindrical fibers frequently formed coiled patterns (Fig. 2b). The length of a single straight segment of a folded zigzag pattern reached up to 7 mm, especially when a large exposed area on the collector was available, i.e. at an early stage of the electrospinning process. The folding of ribbon-like fibers into zigzag patterns was not significantly affected by the substrate (silicon, glass, paper, or aluminum), although there were variations in the length of the pattern segments clamped to the surface. This can be seen in the SEM images in Fig. 2 and 3a (aluminum substrate) and Fig. 3b–d (silicon substrate), where the dark color of the fibers corresponds to their area clamped to the substrate and the lighter color indicates the part of the fiber that does not touch the substrate. An artistic representation of a typical zigzag pattern of the ribbon-like fiber is shown in Fig. 3e.
 | ||
| Fig. 2 SEM images of: (a) PVP/AMT fibers at the early stage of accumulation on an aluminum foil collector, and (b) characteristic patterns formed by ribbon-like (left) and cylindrical (right) fibers. | ||
 | ||
| Fig. 3 SEM images of PVP/AMT folded ribbon-like fibers on (a) aluminum foil and (b–d) silicon wafer; (e) a model of folded ribbon-like fiber structure. | ||
The formation of various coiled and folded polymer fiber patterns due to the buckling of viscous polymer jets has been reported for both uncharged jets directed at and collected on stationary surfaces and electrified jets collected on moving solid surfaces.8,10,38–40 The longitudinal compressive forces resulting from the interaction of jets with the collector have been proven as the cause of buckling. This phenomenon has been shown to occur close to the collector, and has been described in terms of the characteristic frequency of the buckling instability, the wavelength of the formed pattern, and the distance advanced by each cycle, which is proportional to the relative lateral velocity between the oncoming jet and collector. The collector was stationary in the present study. In order to determine similar pattern parameters for the case of PVP/AMT folded zigzag patterns (Fig. 3) prepared from the electrified jet on a stationary surface, the axial jet speed component, vjet, was calculated using the fiber dimensions and volume of dried material collected on the substrate per unit of time. Based on the known flow rate (0.1–0.2 mL h−1) and experimental observation that only a single jet was generated through the process and the material loss was minimal, the average axial jet speed vjet = 17.8 ± 7 m s−1 was calculated. The wavelength λ and the straight segment length S of folded fiber patterns were determined from the SEM images in the ranges of 5–25 μm and 10–50 μm, respectively (Fig. 3a–d). For more than a hundred measured zigzag patterns, the ratio λ/2S varied between 0.15 and 0.6, but there was no apparent relationship established between λ, 2S, and the width of folded ribbon-like structures. Then, the jet lateral speed component relative to the substrate was determined as vlat = (λ/2S)vjet, and the buckling frequency f was found as f = vjet/2S. For the average magnitude of vjet = 17.8 m s−1, the typical lateral speeds and buckling frequencies in the ranges (2.7 ÷ 10.7) m s−1 and (1.8 ÷ 9.0) × 105 Hz were obtained, respectively. The latter values are in reasonable agreement with the results reported for other polymer fiber electrospinning processes.8–10,41,42
Thermal processing of as-prepared PVP/AMT fibrous material at 500 °C in the air led to crystallization of tungsten oxide nanofibers with shapes resembling the original PVP/AMT fibers (Fig. 4). As discussed later in the text, the resulting fibrous tungsten oxide material was identified as a polycrystalline monoclinic WO3 phase with a grain size in the range of 20–100 nm and the level of impurities below the detection limit of the utilized characterization techniques. After annealing, the diameter of the cylindrically-shaped WO3 fibers reduced from the initial 650 ± 200 nm to 250 ± 50 nm. This corresponded to the volume of the crystallized WO3 fiber of about 0.17 of the original volume of PVP/AMT fiber, assuming that the shrinkage along the fiber length was relatively small. The thickness of the resulting tungsten oxide nanoribbons was typically shrunk from the original value of ∼150 nm for PVP/AMT fiber to 80 ± 10 nm, and some porosity developed, but the integrity of the fibrous material remained.
 | ||
| Fig. 4 SEM images of (a and b) loose PVP/AMT ribbon-like fibers, and (c and d) the resulting WO3 nanofibers after annealing for 3 h at 500 °C in the air. | ||
The crystallization of folded zigzag patterns of ribbon-like PVP/AMT fibers had noticeable differences from that of the free-standing fibers (Fig. 5a–d and 6a–c). First, the width of the crystallized WO3 nanoribbon was determined by whether or not the fiber was clamped to the substrate. Second, more non-uniform crystallization and substantial fiber deformation (Fig. 5b and d) was observed in patterns formed by wider fibers and on the substrate with a rougher surface. An artistic representation of a typical zigzag pattern of the annealed ribbon-like WO3 fiber is shown in Fig. 5e. Based on the measurements of ribbon-like fiber widths before and after annealing, it has been concluded that the clamped segments of ribbon-like WO3 fibers closely retain the width of the original PVP/AMT fibers. The shrinkage of the non-clamped segments of the folded zigzag patterns was almost linearly proportional to the width of the clamped sections. For example, for 1.1 μm width of the clamped pattern segment the width of the free-standing part was 0.7 μm, while for 4.0 μm width of the clamped segment the width of the free-standing part was 2.8 μm. Thus, the width of the free-standing segments of ribbon-like fibers shrank, on average, by 35 ± 5%, which was less than the thickness shrinkage (from 150 nm to 80 nm). Since the length of the free-standing segment of the pattern remained nearly the same, the volume of the crystallized WO3 ribbon-like fiber was about 0.35 of the volume of the original PVP/AMT fiber. Thus, the volume of the ribbon-like WO3 fibers (Fig. 6a and b) was about two times larger than that of the cylindrical fibers (Fig. 6c). This difference can be associated with the higher porosity and the rougher surface of the ribbon-like WO3 fibers due to the nearly single grain thickness of the ribbon (Fig. 6d and e).
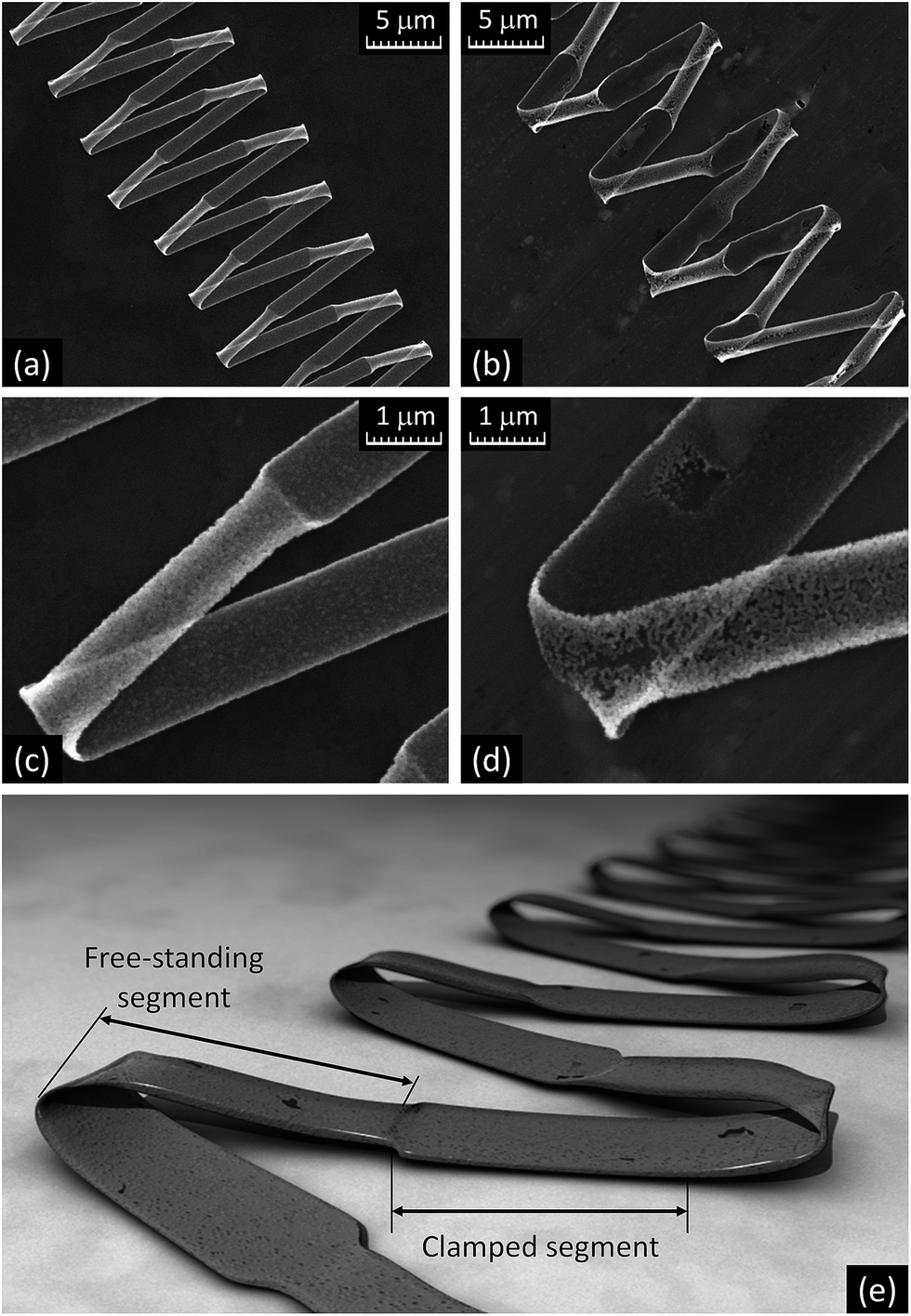 | ||
| Fig. 5 SEM images of folded patterns of WO3 ribbons after the annealing on (a and c) silicon wafer and (b and d) aluminum foil; (e) a model of annealed folded ribbon-like fiber structure. | ||
 | ||
| Fig. 6 (a–c) Comparative SEM images of (a) ribbon-like, (b) folded ribbon-like, and (c) cylindrical WO3 nanofibers; (d and e) representative TEM images of WO3 ribbon-like fibers. | ||
XPS analysis of PVP/AMT ribbon-like fibers (Fig. 7a) yielded the binding energies for the W4f duplet at 35 eV (W4f7/2) and 37 eV (W4f5/2), the O1s level at 532.2 eV (W–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), the C1s level at 284.7 eV (C–C, C–H) and 287.5 eV (CO), and the N1s level at 399.3 eV (N–CO in PVP) and 400.4 eV (N–H in AMT). The W4f duplet was fit with four peaks (33.5 eV and 36.2 eV for W5+, 35.1 eV and 37.2 eV for W6+, respectively), and the W6+/W5+ peak area ratio was 70/30.43 FTIR spectrum of as-prepared PVP/AMT fibers (Fig. 7c, top) revealed three infrared absorption bands with maxima at 933 cm−1 (with a weak shoulder around 959 cm−1), 879 cm−1 and 781 cm−1, which are close to the positions of absorption bands assigned to W–O vibrations in AMT.44 These bands overlap with weak PVP bands in this region of the infrared spectrum. Strong absorption bands at 1643 cm−1 and 1290 cm−1 correspond to CO/C–N stretching and C–H wagging/C–N stretching vibrations in PVP, whereas the bands in 1490–1420 cm−1 region can be assigned to vibrations of C–N and N–CO bonds in PVP and N–H bond in AMT.45,46 These data suggest that tungsten was present mostly in its +6 oxidation state in as-prepared PVP/AMT fibers, and there was little chemical interaction between the WOx polyhedra and PVP matrix. Annealing PVP/AMT fibers in the air for 3 hours at 500 °C resulted in a broad absorption band in FTIR spectrum (Fig. 7c, bottom) with distinguishable spectral features at 1040 cm−1, 922 cm−1, 803 cm−1, 726 cm−1 and 640 cm−1, which fit fairly well the infrared spectrum of a monoclinic stoichiometric WO3 phase.44 The XPS spectrum (Fig. 7b) of the annealed material showed the W4f duplet at 35.1 eV (W4f7/2) and 37.2 eV (W4f5/2), which was fit with only two peaks at the same positions (35.1 eV, FWHM = 1.1 eV and 37.2 eV, FWHM = 1.4 eV) corresponding to W6+, the O1s peaks at 530.2 eV (O2− in W–O) and 532.6 eV (Si–O in the substrate), and a small C1s peak at 285 eV. The latter peak is associated with common surface contamination during sample handling and it disappeared after a 2 minute sputtering with a 500 eV Ar+ ion beam. The calculated composition of the annealed material (based on the entire W4f and 530.2 eV O1s peak areas, and the sensitivity factors of 2.75 for W and 0.66 for O, respectively) corresponded to a sub-stoichiometric WO2.94 in the topmost surface layer of nanofibers.
O), the C1s level at 284.7 eV (C–C, C–H) and 287.5 eV (CO), and the N1s level at 399.3 eV (N–CO in PVP) and 400.4 eV (N–H in AMT). The W4f duplet was fit with four peaks (33.5 eV and 36.2 eV for W5+, 35.1 eV and 37.2 eV for W6+, respectively), and the W6+/W5+ peak area ratio was 70/30.43 FTIR spectrum of as-prepared PVP/AMT fibers (Fig. 7c, top) revealed three infrared absorption bands with maxima at 933 cm−1 (with a weak shoulder around 959 cm−1), 879 cm−1 and 781 cm−1, which are close to the positions of absorption bands assigned to W–O vibrations in AMT.44 These bands overlap with weak PVP bands in this region of the infrared spectrum. Strong absorption bands at 1643 cm−1 and 1290 cm−1 correspond to CO/C–N stretching and C–H wagging/C–N stretching vibrations in PVP, whereas the bands in 1490–1420 cm−1 region can be assigned to vibrations of C–N and N–CO bonds in PVP and N–H bond in AMT.45,46 These data suggest that tungsten was present mostly in its +6 oxidation state in as-prepared PVP/AMT fibers, and there was little chemical interaction between the WOx polyhedra and PVP matrix. Annealing PVP/AMT fibers in the air for 3 hours at 500 °C resulted in a broad absorption band in FTIR spectrum (Fig. 7c, bottom) with distinguishable spectral features at 1040 cm−1, 922 cm−1, 803 cm−1, 726 cm−1 and 640 cm−1, which fit fairly well the infrared spectrum of a monoclinic stoichiometric WO3 phase.44 The XPS spectrum (Fig. 7b) of the annealed material showed the W4f duplet at 35.1 eV (W4f7/2) and 37.2 eV (W4f5/2), which was fit with only two peaks at the same positions (35.1 eV, FWHM = 1.1 eV and 37.2 eV, FWHM = 1.4 eV) corresponding to W6+, the O1s peaks at 530.2 eV (O2− in W–O) and 532.6 eV (Si–O in the substrate), and a small C1s peak at 285 eV. The latter peak is associated with common surface contamination during sample handling and it disappeared after a 2 minute sputtering with a 500 eV Ar+ ion beam. The calculated composition of the annealed material (based on the entire W4f and 530.2 eV O1s peak areas, and the sensitivity factors of 2.75 for W and 0.66 for O, respectively) corresponded to a sub-stoichiometric WO2.94 in the topmost surface layer of nanofibers.
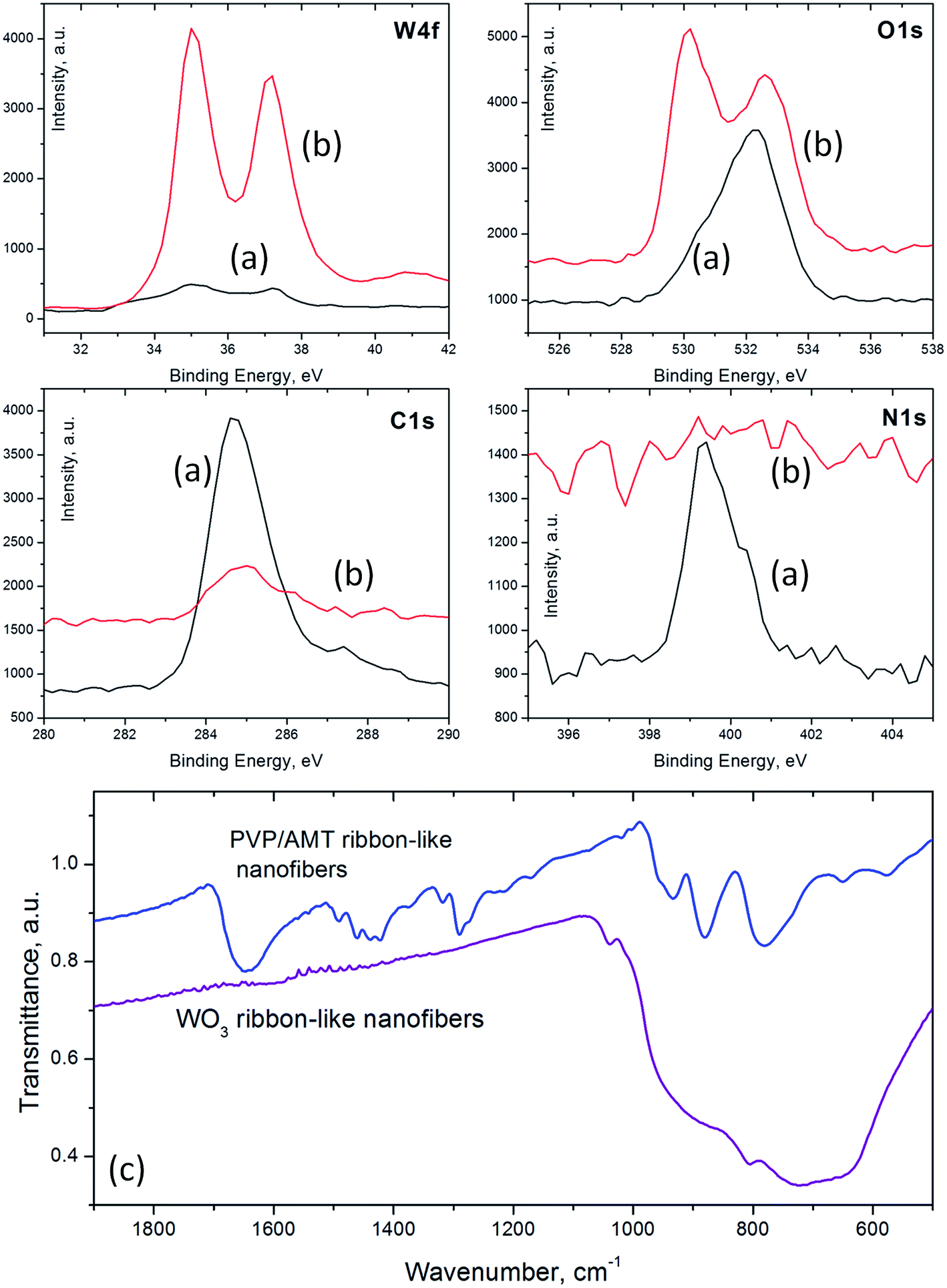 | ||
| Fig. 7 XPS (W4f, C1s, O1s, and N1s) spectra of (a) as prepared PVP/AMT ribbon-like nanofibers and (b) the resulting WO3 fibers after thermal processing for 3 h at 500 °C in the air; (c) FTIR spectra of PVP/AMT (top) and annealed WO3 (bottom) fibers. | ||
X-ray diffraction spectra (Fig. 8a) recorded for thin layers of predominantly ribbon-like WO3 fibers and their folded patterns showed peaks located with an accuracy of ±0.1° at 23°, 23.6°, 24.3°, 26.5°, 28.6°, 31°, 32.9°, 33.3°, 34°, 41.6°, 44°, 45.7°, 48.3°, and 50°. The positions of these peaks closely match either the monoclinic or triclinic WO3 phase. On the other hand, Raman spectra recorded for the same samples (Fig. 8b) showed the peaks located at 137 cm−1, 193 cm−1 (lattice modes), 273 cm−1, 330 cm−1 (O–W–O deformation vibrations), 522 cm−1 (Si), as well as 714 cm−1, 808 cm−1, and a weak broad band from 942 cm−1 to 980 cm−1. The 714 cm−1 and 808 cm−1 Raman bands correspond to stretching vibrations of bridging oxygen in O–W–O.47 The scattering between 942 cm−1 and 980 cm−1 can originate due to the presence of WO terminal modes of surface grains.48 The Raman bands at 610 cm−1 and 241 cm−1 associated with triclinic WO3 phase were not observed.49
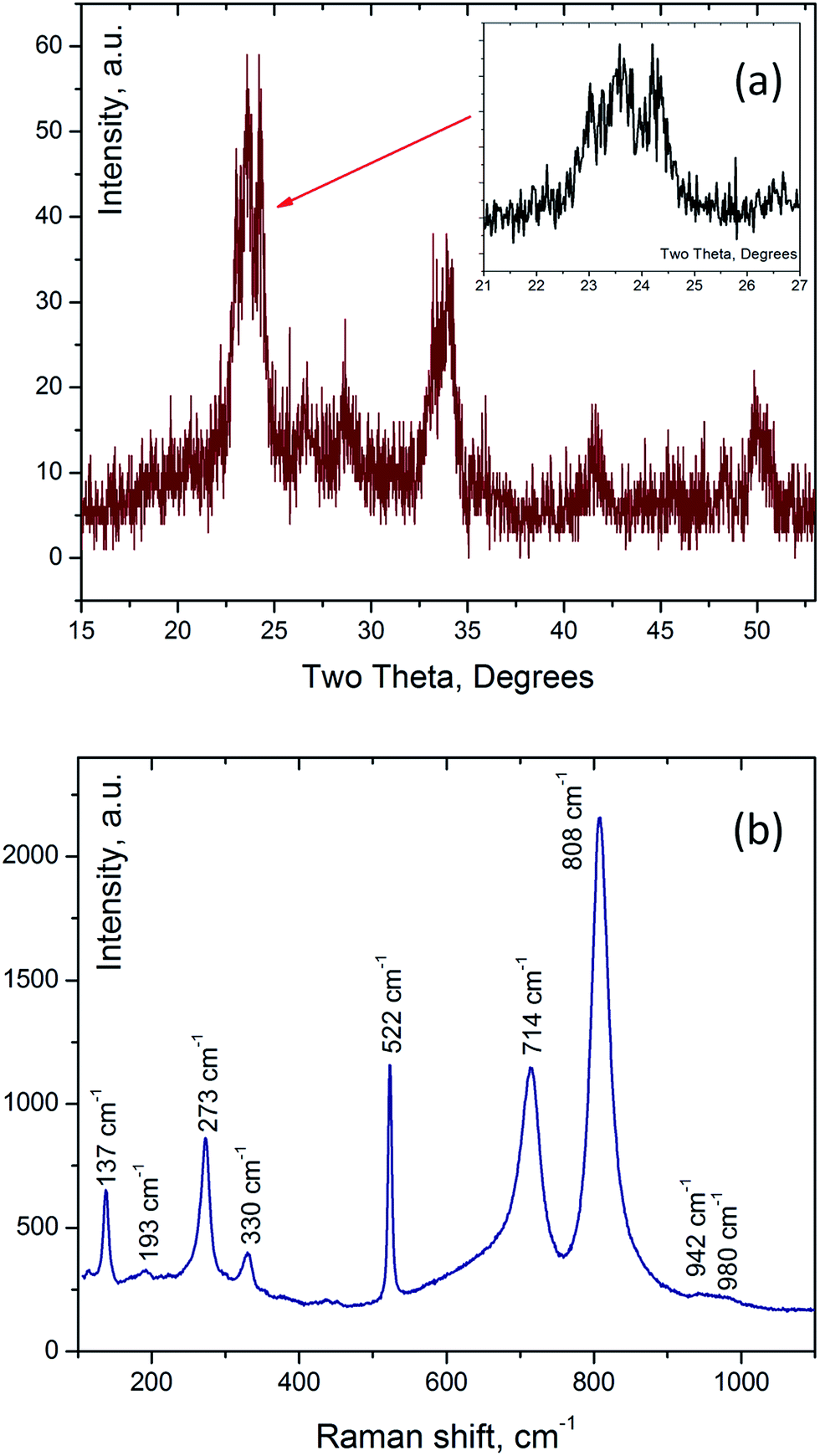 | ||
| Fig. 8 (a) X-ray diffraction pattern and (b) Raman (excitation wavelength 532 nm) spectrum of a thin layer of ribbon-like WO3 nanofibers on silicon substrate. | ||
Conclusions
It has been demonstrated that a large yield of uncommonly shaped ribbon-like PVP/AMT fibers can be achieved with a suitable combination of the parameters of a capillary needle dc-electrospinning process. However, the parameter window for the generation of predominantly ribbon-like fibers seems to be rather narrow. The concentrations and associated properties (viscosity, surface tension, electrical conductivity, etc.) of the precursor needed for the generation of ribbon-like fibers may be beyond the precursor spinnability threshold for a particular method of electrospinning.When collected on a smooth stationary substrate, zigzag patterns of folded ribbon-like fibers formed at the early stage of the dc-electrospinning process. This phenomenon seemed to be mainly governed by the same physical principles as in the case of non-charged jets or charged polymer jets depositing on the moving collector.
Annealing of the fabricated ribbon-like PVP/AMT fibers and folded fiber patterns at 500 °C in the air led to the formation of polycrystalline monoclinic phase WO3 structures with shapes similar to the original PVP/AMT fibers. For the crystallized ribbon-like WO3 fibers, the typical thickness was 80 ± 10 nm and the width-to-thickness ratio was up to 50:1. Although the shape of the original PVP/AMT fibers was generally preserved in WO3 fibers, the ribbon-like WO3 fibers demonstrated higher porosity and noticeably lower mechanical stability when compared to cylindrical fibers.
Acknowledgements
This work has been supported in part by the National Science Foundation International Research Experience for Students (IRES) award to UAB (Grant #1261154). SEM and XPS instrumentation used in this work have been supported by the NSF DMR Grants #1040474 and #0922910, respectively.References
- T. R. Hayes and I. C. Hosie, Nanosci. Nanotechnol., 2015, 96, 305–329 Search PubMed.
- Y. Dai, W. Liu, E. Formo, Y. Sun and Y. Xia, Polym. Adv. Technol., 2011, 22, 326–338 CrossRef CAS PubMed.
- W. E. Teo, R. Inai and S. Ramakrishna, Sci. Technol. Adv. Mater., 2011, 12, 013002 CrossRef.
- H. Wu, W. Pan, D. Lin and H. Li, J. Adv. Ceram., 2012, 1, 2–23 CrossRef CAS.
- I. D. Kim and A. Rothschild, Polym. Adv. Technol., 2011, 22, 318–325 CrossRef CAS PubMed.
- X. Zhang, L. Ji, Z. Lin and Y. Li, Adv. Mater. Res., 2012, 441, 231–234 CrossRef CAS.
- K. Garg and G. L. Bowlin, Biomicrofluidics, 2011, 5, 013403 CrossRef PubMed.
- S. Koombhongse, W. Liu and D. H. Reneker, J. Polym. Sci., Part B: Polym. Phys., 2001, 39, 2598–2606 CrossRef CAS PubMed.
- D. H. Reneker and A. L. Yarin, Polymer, 2008, 49, 2387–2425 CrossRef CAS PubMed.
- T. Han, D. H. Reneker and A. L. Yarin, Polymer, 2007, 48, 6064–6076 CrossRef CAS PubMed.
- Q. Lu, S. Liu, M. Ren, L. Song and G. Zhao, J. Sol-Gel Sci. Technol., 2012, 61, 169–174 CrossRef CAS.
- J. T. McCann, J. I. L. Chen, D. Li, Z. G. Ye and Y. Xia, Chem. Phys. Lett., 2006, 424, 162–166 CrossRef CAS PubMed.
- S. Zhang, X. Y. Cao and Y. M. Ma, J. Optoelectron. Adv. Mater., 2011, 13, 425–427 CAS.
- S. Li, Z. He, X. Wang and K. Gao, Appl. Phys. A: Mater. Sci. Process., 2014, 117, 1381–1386 CrossRef CAS.
- M. Ristić, M. Marciuš, M. Ž. Petrović, M. Ivanda and S. Musić, Mater. Lett., 2015, 156, 142–145 CrossRef PubMed.
- G. Zhao, S. Liu, Q. Lu, F. Xu and H. Sun, J. Alloys Compd., 2013, 578, 12–16 CrossRef CAS PubMed.
- S. J. Choi, C. Choi, S. J. Kim, H. J. Cho, M. Hakim, S. Jeon and I. D. Kim, Sci. Rep., 2015, 5, 8067 CrossRef CAS PubMed.
- J. H. Song, J. H. Nam, J. H. Cho, B. I. Kim, M. P. Chun and D. K. Choi, J. Korean Phys. Soc., 2011, 59, 2308–2312 CrossRef CAS.
- F. K. Butt, C. Cao, W. S. Khan, M. Safdar, X. Fu, M. Tahir, F. Idrees, Z. Ali, G. Nabi and D. Yu, CrystEngComm, 2013, 15, 2106–2112 RSC.
- Z. Hou, H. Lian, M. Zhang, L. Wang, M. Lü, C. Zhang and J. Lin, J. Electrochem. Soc., 2009, 156, J209–J214 CrossRef CAS PubMed.
- Y. Su, B. Lu, Y. Xie, Z. Ma, L. Liu, H. Zhao, J. Zhang, H. Duan, H. Zhang, J. Li, Y. Xiong and E. Xie, Nanotechnology, 2011, 22, 285609 CrossRef PubMed.
- J. Muangban and P. Jaroenapibal, Ceram. Int., 2014, 40, 6759–6764 CrossRef CAS PubMed.
- T. A. Nguyen, S. Park, J. B. Kim, T. K. Kim, G. H. Seong, J. Choo and Y. S. Kim, Sens. Actuators, B, 2011, 160, 549–554 CrossRef CAS PubMed.
- X. Lu, X. Liu, W. Zhang, C. Wang and Y. Wei, J. Colloid Interface Sci., 2006, 298, 996–999 CrossRef CAS PubMed.
- Á. K. Nagy, J. Pfeifer, I. E. Lukács, A. L. Tóth and C. Balázsi, Mater. Sci. Forum, 2010, 659, 215–219 CrossRef.
- I. M. Szilágyi, E. Santala, M. Heikkilä, M. Kemell, T. Nikitin, L. Khriachthev, M. Räsänen, M. Ritala and M. Leskelä, J. Therm. Anal. Calorim., 2011, 105, 73–81 CrossRef.
- S. Piperno, M. Passacantando, S. Santucci, L. Lozzi and S. La Rosa, J. Appl. Phys., 2007, 101, 124504 CrossRef PubMed.
- G. Wang, Y. Ji, X. Huang, X. Yang, P. I. Gouma and M. Dudley, J. Phys. Chem. B, 2006, 110, 23777–23782 CrossRef CAS PubMed.
- J. Y. Leng, X. J. Xu, N. Lv, H. T. Fan and T. Zhang, J. Colloid Interface Sci., 2011, 356, 54–57 CrossRef CAS PubMed.
- W. H. Lai, J. Shieh, L. G. Teoh and M. H. Hon, Nanotechnology, 2006, 17, 110–115 CrossRef CAS.
- X. Chi, C. Liu, L. Liu, Y. Li, Z. Wang, X. Bo, L. Liu and C. Su, Sens. Actuators, B, 2014, 194, 33–37 CrossRef CAS PubMed.
- Z. Chen, W. Wang and K. Zhu, Acta Metall. Sin., 2014, 28, 1–6 CrossRef.
- H. S. Shim, J. W. Kim, Y. E. Sung and W. B. Kim, Sol. Energy Mater. Sol. Cells, 2009, 93, 2062–2068 CrossRef CAS PubMed.
- P. Pokorny, E. Kostakova, F. Sanetrnik, P. Mikes, J. Chvojka, T. Kalous, M. Bilek, K. Pejchar, J. Valtera and D. Lukas, Phys. Chem. Chem. Phys., 2014, 16, 26816–26822 RSC.
- O. Jirsak and S. Petrik, Int. J. Nanotechnol., 2012, 9, 836–845 CrossRef CAS.
- Powder Diffraction File, ed. S. Kabekkodu, ICDD, 2007 Search PubMed.
- F. L. Zhou, R. H. Gong and I. Porat, J. Appl. Polym. Sci., 2010, 115, 2591–2598 CrossRef CAS PubMed.
- S. Chiu-Webster and J. R. Lister, J. Fluid Mech., 2006, 569, 89–111 CrossRef.
- N. M. Ribe, J. R. Lister and S. Chiu-Webster, Phys. Fluids, 2006, 18, 124105 CrossRef PubMed.
- L. Huang, R. A. McMillan, R. P. Apkarian, B. Pourdeyhimi, V. P. Conticello and E. L. Chaikof, Macromolecules, 2000, 33, 2989–2997 CrossRef CAS.
- I. Greenfeld, K. Fezzaa, M. H. Rafailovich and E. Zussman, Macromolecules, 2012, 45, 3616–3626 CrossRef CAS.
- L. M. Bellan, H. G. Craighead and J. P. Hinestroza, J. Appl. Phys., 2007, 102, 094308 CrossRef PubMed.
- I. M. Szilágyi, F. Hange, J. Madarász and G. Pokol, Eur. J. Inorg. Chem., 2006, 17, 3413–3418 CrossRef PubMed.
- L. M. Plyasova, I. Y. Molina, G. N. Kustova, G. S. Litvak, A. I. Nizovskii and A. N. Gavrilov, J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech., 2009, 3, 869–875 CrossRef.
- T. A. Nguyen, T. S. Jun, M. Rashid and Y. S. Kim, Mater. Lett., 2011, 65, 2823–2825 CrossRef CAS PubMed.
- Y. Borodko, S. E. Habas, M. Koebel, P. Yang, H. Frei and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 23052–23059 CrossRef CAS PubMed.
- M. F. Daniel, B. Desbat, J. C. Lassegues, B. Gerand and M. Figlarz, J. Solid State Chem., 1987, 67, 235–247 CrossRef CAS.
- J. Pfeifer, C. Guifang, P. Tekula-Buxbaum, B. A. Kiss, M. Farkas-Jahnke and K. Vadasdi, J. Solid State Chem., 1995, 119, 90–97 CrossRef CAS.
- G. N. Kustova, Y. A. Chesalov, L. M. Plyasova, I. Y. Molina and A. I. Nizovskii, Vib. Spectrosc., 2011, 55, 235–240 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |