
Jatrorrhizine hydrochloride potentiates the neuraminidase inhibitory effect of oseltamivir towards H7N9 influenza
Abstract
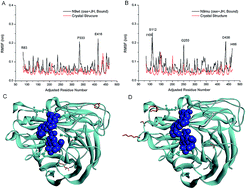
A Chinese natural product, jatrorrhizine hydrochloride (JH), exhibited a potent inhibitory effect toward neuraminidase of the H7N9 (N9) avian influenza virus. The interaction between JH and N9 was modeled using the molecular docking software AutoDock. Similar to the control drug oseltamivir, JH bound into the active site of N9, but its position did not overlap with the position of oseltamivir. In addition, JH and oseltamivir were simultaneously docked into the active site of N9s (N9WT and N9R294K). By analyzing the docking result of the binding position, hydrogen bond interaction, and pi–pi interaction, N9 inhibition was likely enhanced by the combination of JH and oseltamivir. Molecular dynamics simulations using GROMACS were used to obtain trajectories of N9s and two compound complexes (JH was chosen from the docking result, whereas oseltamivir was directly extracted from the crystal structure) and to validate key interactions. The coordination between JH and oseltamivir to inhibit neuraminidase activity was further confirmed by the enzymatic assays of N9s. This study introduces novel treatment strategies to target N9 for a potential anti-H7N9 influenza drug.
 Please wait while we load your content...
Please wait while we load your content...

