
Nickel-promoted mesoporous ZSM5 for carbon monoxide methanation

Abstract
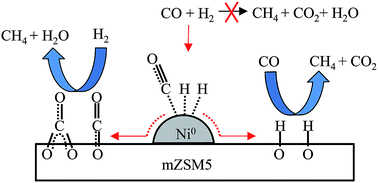
Nickel-promoted mesoporous ZSM5 (Ni/mZSM5) was prepared for CO methanation. XRD, NMR and SEM analysis confirmed the structural stability of Ni/mZSM5 with coffin type morphology. The nitrogen physisorption and pyrrole adsorbed FTIR analyses indicated the presence of micro–mesoporosity and a moderate amount of basic sites on both mZSM5 and Ni/mZSM5. At 623 K, Ni/mZSM5 showed a high rate of CO conversion (141.6 μmol CO g-cat−1 s−1) and 92% CH4 yield. Ni/mZSM5 showed better catalytic performance than Ni/MSN (82.4 μmol CO g-cat−1 s−1, 82% CH4 yield), Ni/HZSM5 (29.0 μmol CO g-cat−1 s−1, 54.5% CH4 yield), and Ni/γ-Al2O3 (14.5 μmol CO g-cat−1 s−1, 38.6% CH4 yield). It is noteworthy that the superior catalytic performance of Ni/mZSM5 could be attributed to the presence of both micro–mesoporosity and basicity, which led to a synergistic effect of Ni metal active sites and the mZSM5 support. In situ FTIR spectroscopy showed that CO and H2 may be adsorbed on Ni metal followed by spillover to form adsorbed CO and adsorbed H on the mZSM5 surface. Then, two possible mechanisms for CO methanation were proposed. In the first mechanism, the adsorbed CO may be reacted with H2 to form CH4 and H2O. In the second mechanism, the adsorbed H may be reacted with CO to form CH4 and CO2. However, in this case, the former is the predominant pathway as the methanation reaction is favored by inhibition of the water–gas shift reaction.
 Please wait while we load your content...
Please wait while we load your content...

