
Sonochemical synthesis of cyclophosphazene bridged mesoporous organosilicas and their application in methyl orange, congo red and Cr(vi) removal†
Abstract
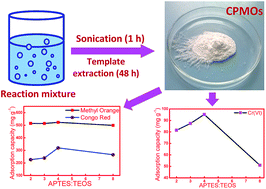
A rapid sonochemical route was adopted for the synthesis of cyclophosphazene bridged mesoporous organosilicas (CPMOs) by co-condensation of (3-aminopropyl)triethoxysilane (APTES) and phosphonitrilic chloride trimer (PNC) with variable amounts of tetraethyl orthosilicate (TEOS) in the presence of cetyltrimethylammonium bromide (CTABr). The products were formed as early as 15 min, however, the experiments were carried out for 1 h for better condensation and aging. The specific surface areas of the specimens were a function of the APTES to TEOS ratio and varied in the range of 58 to 974 m2 g−1. The pore size distribution of these specimens was centred around 1.4 to 3.7 nm. These CPMOs were employed for the adsorption of organic dyes such as methyl orange (MO) and congo red (CR) as well as Cr(VI) ions. Adsorption studies were carried out in aqueous solution by varying the contact time, initial dye concentration, and temperature. The equilibrium data were fitted using the Langmuir and Freundlich isotherm by linear regression analysis. The kinetic analysis revealed that the overall adsorption process was pseudo-second-order. The maximum adsorption capacities were 523 mg g−1, 320 mg g−1 and 101 mg g−1 for MO, CR and Cr(VI) ions, respectively at 25 °C. The adsorption was a spontaneous exothermic process with negative ΔG and ΔH derived from the thermodynamics studies.
 Please wait while we load your content...
Please wait while we load your content...

