
Selective protein purification by PEG–IDA-functionalized iron oxide nanoparticles†
Abstract
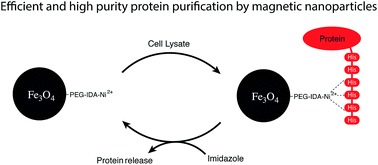
We developed a new heterobifunctional polyethylene glycol ligand for iron oxide nanoparticles with a group for covalent surface attachment and for chelating metal ions. After introduction of nickel ions, His-tagged fluorescent proteins were magnetically purified from a cell lysate. A high purity product and efficient magnetic separation were observed.
 Please wait while we load your content...
Please wait while we load your content...

