
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of counter ions on the far-infrared spectra of tris(triphenylphosphinegold)oxonium dimer salts†
Trystan
Bennett
a,
Alexander J.
Falcinella
a,
Reuben J.
White
a,
Rohul H.
Adnan
bc,
Vladimir
Golovko
*b,
Gunther G.
Andersson
*d and
Gregory F.
Metha
*a
aDepartment of Chemistry, University of Adelaide, South Australia 5005, Australia. E-mail: greg.metha@adelaide.edu.au; Fax: +61 8 8313 4358; Tel: +61 8 8313 5943
bThe MacDiarmid Institute for Advanced Materials and Nanotechnology, Department of Chemistry, University of Canterbury, Christchurch 8140, New Zealand
cChemistry Department, University of Malaya, 50603 Kuala Lumpur, Malaysia
dFlinders Centre for NanoScale Science and Technology, Flinders University, Adelaide, SA 5001, Australia
First published on 2nd September 2015
Abstract
Two tris(triphenylphosphinegold)oxonium dimer salts [{{Au(PPh3)}3(μ3−O)}2]2+(X−)2 (X = BF−4, MnO−4) were investigated via synchrotron-based far-infrared vibrational spectroscopy and density functional theory modelled at the M06/LANL2DZ level of theory. The 50–800 cm−1 region of both oxonium salts is presented, with the spectrum for [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 found to possess a large feature at 330.3 cm−1, attributable to counter-ion vibrational modes, which is only predicted upon explicit inclusion of counter-ions in the calculation. A feature around 107 cm−1 observed for the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 infrared spectrum is assigned to 21 distinct vibrational modes arising from Au–Au bond stretching and other motions of the Au core. The same feature is predicted to be present within the [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 spectrum but is masked by experimental noise. In the 50–400 cm−1 region, the relative intensities of predicted vibrational modes is found to depend heavily on the presence and nature of the counter-ions, while within the 400–800 cm−1 region, little dependence of the theoretical spectra on the type of counter-ion is predicted. Finally, the dimerization energies of both [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 are calculated to be 3.06 eV and 3.20 eV, respectively, when the counter-ions are explicitly included within the calculation, and just 1.10 eV in their absence.
Introduction
The chemistry of univalent gold species has been well studied throughout the past 40 years,1 spurred primarily by the work of Nesmeyanov and co-workers.2–7 The discovery of the tris(triphenylphosphinegold)oxonium salts of the form [(Ph3PAu)3O]X (X = BF−4, MnO−4, CF3COO−), which have been shown to exist as dimers in the crystalline phase, represented an addition to the few known species which incorporate direct Au–O bonding.2,3 These oxonium salts have seen employment throughout the literature due to their ability to readily decompose in situ into highly reactive Ph3PAu+ species,7 capable of reacting with a wide array of organic species to yield corresponding aurated derivatives. Oxonium salts have been reported to react with cyclic structures such as cyclopentadiene, tetraphenylcyclopentadiene, and ferrocene, resulting in the substitution of H by AuPPh3.5,6,8,9 They have been reported to react with carbon-containing species such as methyl ketones, vinyl esters and ethers, and chloroform, yielding α-aurated species such as aurated acetone, aurated acetaldehyde, and trichloromethyl gold.7 Tris(triphenylphosphinegold)oxonium salts have been shown to react with nitrogen-containing compounds such as malononitrile, N,N-diethylamides, and dicyanoacetylene to yield gold triphenylphosphine-bound species.2,4,7,10–12 The oxonium salts have also been reported to form mixed-metal ligated clusters, with, for example, the species Au3CoRu3(CO)12(PPh3)3 having been synthesized utilizing tris(triphenylphosphinegold)oxonium species as a reagent.13,14Infrared vibrational spectroscopy is a standard technique for probing the unique vibrational fingerprint of a given molecule, which is widely utilized to identify functional groups present within a molecule of interest. For the tris(triphenylphosphategold)oxonium salts, only a rudimentary assignment of the infrared absorption features have been reported,3 with no reports of spectra in the far-infrared region below 800 cm−1. This region is of importance to ligated metal cluster species as vibrational modes involving metal–metal stretching appear within this region.15,16 We have previously reported the far-infrared (50–650 cm−1) vibrational spectra of a range of gold, ruthenium, and gold–ruthenium species, which were accompanied by computational investigations to give an insight into the specific vibrational modes which give rise to each feature within the infrared spectra.15,16 For all previous calculations involving species with counter-ions, the counter-ions were removed and the charge balanced for the primary cluster of interest, i.e. the counter-ions were not explicitly calculated.
The present work reports the vibrational spectra of the oxonium salts [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2, and utilize density functional theory (DFT) based calculations to assign the infrared features of each of these species to specific molecular vibrational modes. Herein, we demonstrate the importance of including counter-ions within the calculations to explain all observed features within the experimental spectrum. As well, the energies of association of the counter-ions to the gold oxonium species have also been calculated, and are reported. Finally, consistent with literature reports that the salts exist as dimers,3,7 the energy of dimerization for both oxonium salts are calculated and presented.
Experimental & computational procedures
The [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 clusters were prepared according to the procedures reported by Nesmeyanov et al.3 Samples were prepared by pressing varying amounts of cluster into polyethylene (PE) to form a pellet of 7 mm diameter. Due to the wide range of absorptivity that the clusters exhibited across the measurement window, the spectra were recorded at several dilutions by mass in PE. For [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2, the spectra reported were recorded with a pure pellet of cluster (solid line in Fig. 1a) and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 cluster:PE (solid line in Fig. 1b) dilution by mass. For [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2, the spectra reported were recorded at 1:1 cluster:PE (solid line in Fig. 2a) and 1:3 cluster:PE (solid line in Fig. 2b) dilutions by mass.
:3 cluster:PE (solid line in Fig. 1b) dilution by mass. For [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2, the spectra reported were recorded at 1:1 cluster:PE (solid line in Fig. 2a) and 1:3 cluster:PE (solid line in Fig. 2b) dilutions by mass.
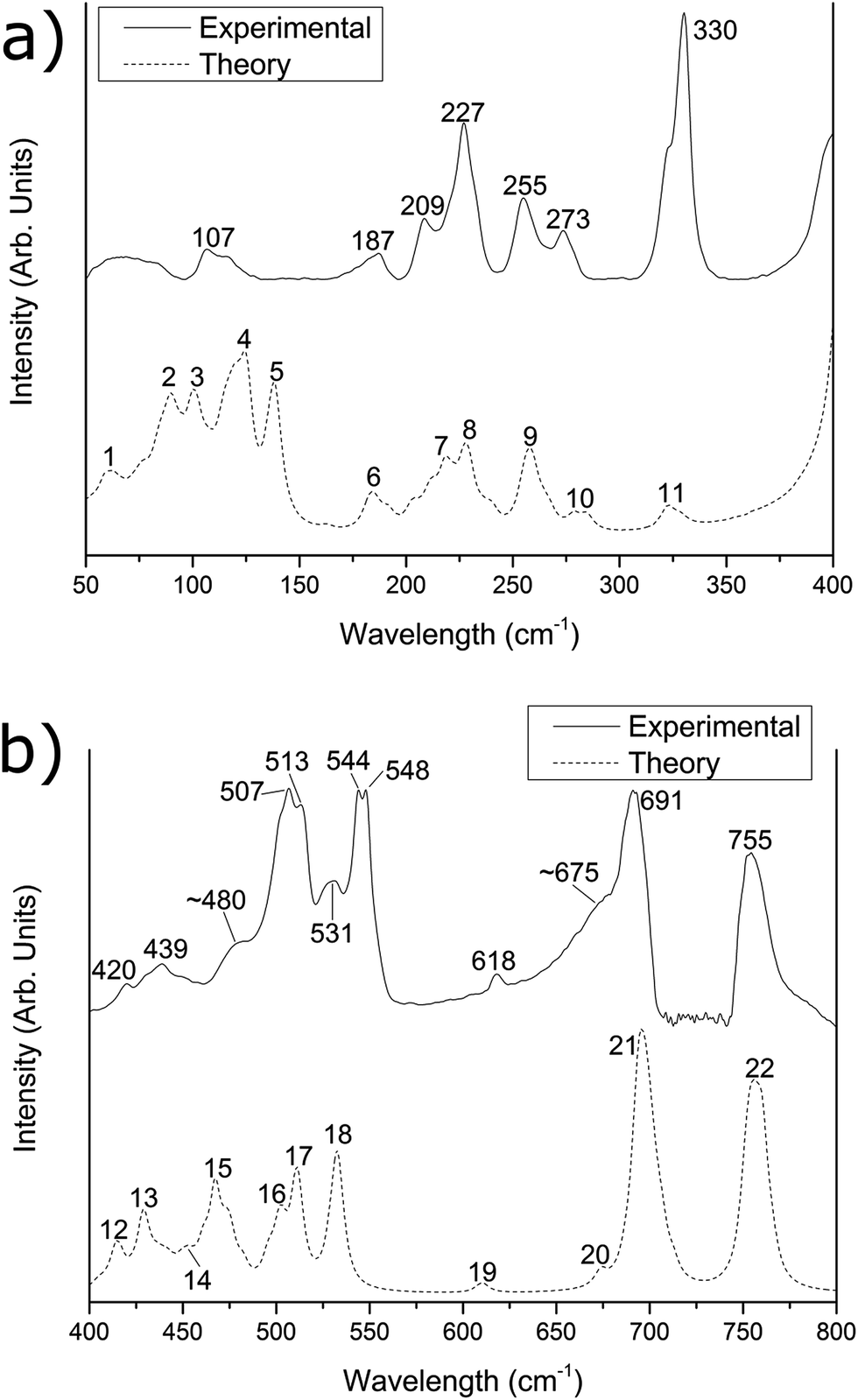 | ||
| Fig. 1 Experimental (solid line) and simulated (dashed line) far-IR spectra of the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 cluster in the (a) 50–400 cm−1 region and (b) 400–800 cm−1 region. The experimental data was recorded at (a) pure sample and (b) a 1:3 sample/PE dilution by mass. The simulated spectrum has been corrected by eqn (1). | ||
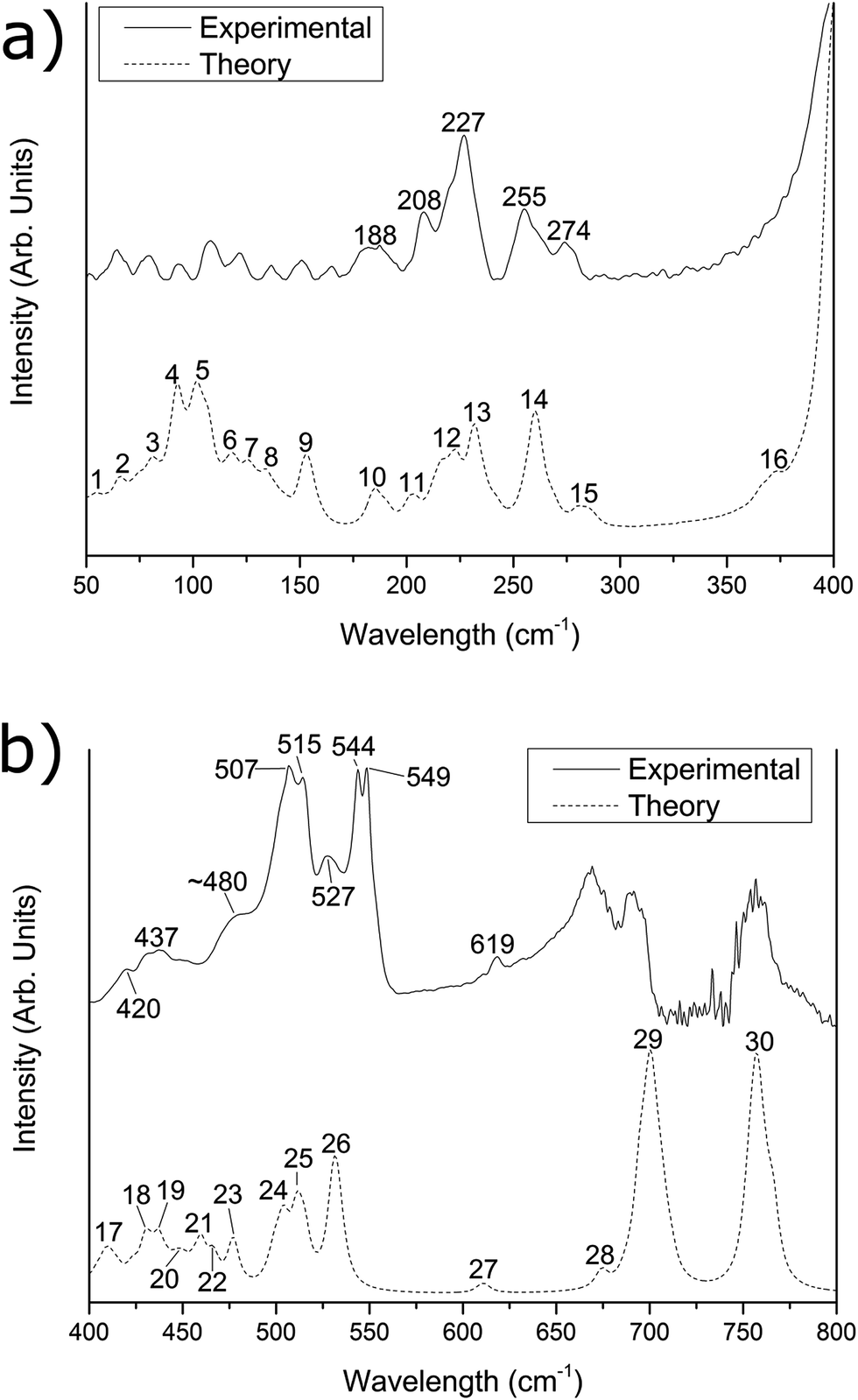 | ||
| Fig. 2 Experimental (solid line) and simulated (dashed line) far-IR spectra of the [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 cluster in the (a) 50–400 cm−1 region and (b) 400–800 cm−1 region. The experimental data was recorded at (a) a 1:1 sample/PE dilution by mass and (b) a 1:3 dilution by mass. The simulated spectrum has been corrected by eqn (1). | ||
The far-IR absorption spectra were recorded using the IFS125 Bruker FT spectrometer located at the far-IR beamline, at the Australian Synchrotron. The transmission spectrum for each sample was recorded from 50 to 800 cm−1, at 1 cm−1 resolution utilizing the synchrotron light source (200 mA in top-up mode), with a 6 micron thick multilayer Mylar beamsplitter in combination with a Si bolometer detector. This bolometer was equipped with an 800 cm−1 far-IR cut-on cold-filter consisting of a 13 micron PE film overlaid with a 6 micron diamond scatter layer. All spectra were recorded at room temperature, and have been baseline corrected.
Geometry optimization and harmonic vibrational frequency calculations of the [{{Au(PPh3)}3(μ3−O)}2]2+(X−)2 (X = BF−4, MnO−4) cluster compounds, including all ligands, were undertaken using the M06 density functional,17 with all atoms treated using the LANL2DZ basis set and related Effective Core Potentials (ECP)18–21 in the Gaussian 09 suite of programs.22 This level of theory has been utilized by us for similar ligated metal cluster systems previously, and has been shown to give good agreement to experimental results.15,16 Except where calculating the energy of dimerization, all calculations reported are for the dimer of the Au3 species, i.e. [{{Au(PPh3)}3(μ3−O)}2]2+(X−)2, (X = BF−4, MnO−4, or nothing), consistent with literature reports.3,7 The starting geometry of the [{{Au(PPh3)}3(μ3−O)}2]2+ species without counter-ions were taken from the X-ray crystal structure of the [{{Au(PPh3)}3(μ3−O)}2]2+ cluster with a N-(4-methoxyphenyl)-2,4,6-trinitroanilate counter-ion (CSD Refcode WAVWUM)23 retrieved from the Cambridge Crystallographic Database.24 The counter-ions and solvent molecules were removed from this structure, and electrons removed from the dimer to balance the charge; a comparison between the X-ray crystal structure and the final optimized gas-phase structure is shown in the ESI (Fig. S1†). All calculations involving binding energies and energies of dimerization were not corrected for Basis Set Superposition Error (BSSE).
Once the [{{Au(PPh3)}3(μ3−O)}2]2+ structure was optimized, the respective counter-ions (either BF−4 or MnO−4) were added and this structure was further optimized at the same level of theory. Several potential starting counter-ion positions were investigated, and for each species the global minimum structure was utilized for frequency calculations and analysis. The charge was explicitly held on each molecule within all calculations which included counter-ions, i.e. the BF−4 or MnO−4 was explicitly held at −1 formal charge, and the [{{Au(PPh3)}3(μ3−O)}2]2+ species was held at +2 formal charge. All calculations were performed as closed-shell species. The optimizations were all performed in the C1 point group, with no potential symmetry identified for all systems.
The calculations were carried out using a larger than default grid (Gaussian keyword “int = ultrafine”) for numerical integral evaluation, with all other cut-offs being left at the default. Each geometry optimization was followed by a harmonic frequency calculation to confirm that the geometry was a true minimum with no imaginary frequencies. To obtain the predicted IR spectra, each predicted stick spectrum was convoluted with a Gaussian line shape function with 8 cm−1 full width at half maximum using the GaussView 5 program, to best match the experimental spectra. Full geometric information for each optimized structure is provided in the accompanying ESI.†
Results & discussion
The predicted spectra over the range 50–800 cm−1 for [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 are shown in Fig. 1 and 2 as the lower dotted lines. The assignments of peaks observed in the experimental spectra to the theoretically predicted spectra for both clusters are shown in Table 1, along with a breakdown of the major vibrational motions for each feature. These spectra only include the calculated fundamental vibrations and do not include overtones or combination bands. For both Fig. 1 and 2, as well as all values in Table 1, the theoretical infrared spectrum has been scaled by the functional-independent correction factor (eqn (1)) reported by Fielicke and co-workers for low-frequency vibrations predicted by DFT calculations.25 This has been used by us previously to obtain better agreement for far-IR transitions than traditional vibrational frequency correction schemes.16![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) corr = 22 cm−1 + calc × 0.94 corr = 22 cm−1 + calc × 0.94 | (1) |
| [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 | [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Freq (cm−1) | Assignment | # Contributing modes | Mode description | % Weighted contributiona | Freq (cm−1) | Assignment | # Contributing modes | Mode description | % Weighted contributiona | ||
| Core (Au/O) | Counter-ions | Core (Au/O) | Counter-ions | ||||||||
| a For each feature in the experimental spectrum, this is the average contribution by the Au/O core atoms toward each vibrational mode, weighted by the predicted infrared activity. b This feature was not able to be resolved due to experimental noise. c This feature was not observed, possibly due to occlusion from neighbouring features (see text), however is included for comparison. d These features are not assigned due to poorly resolved peaks, due to their proximity to the polyethylene window (see text). | |||||||||||
| 107 | 4 | 13 | Au core motion | 10.6% | 12.5% | b | 4/5 | 20 | Au core motion | 6.0% | 11.3% |
| 187 | 6 | 6 | Au–P stretching | 10.3% | 2.1% | 188 | 10 | 6 | Au–P stretching | 10.1% | 2.5% |
| 209 | 7 | 10 | Ph3P group rotating | 3.0% | 0.9% | 208 | 11/12 | 10 | Ph3P group rotating | 2.9% | 1.2% |
| 227 | 8 | 6 | Ph group rocking | 1.1% | 1.7% | 227 | 13 | 6 | Ph group rocking | 1.1% | 2.1% |
| 255 | 9 | 12 | Ph–P–Ph bending | 0.9% | 0.7% | 255 | 14 | 12 | Ph–P–Ph bending | 0.8% | 0.3% |
| 273 | 10 | 6 | Ph–P–Ph bending | 0.1% | 1.1% | 274 | 15 | 6 | Ph–P–Ph bending | 0.3% | 0.6% |
| 330 | 11 | 4 | BF4 bending and scissoring | 0.8% | 79.5% | c | 16 | 4 | MnO4 bending motion | 2.7% | 75.7% |
| 420 | 12 | 19 | Core O motion; Ph group out-of-plane distortions | 6.9% | 0.6% | 420 | 17 | 22 | Core O motion; coupled to C–I scissoring modes; Ph group out-of-plane distortions | 6.6% | 16.0% |
| 439 | 13 | 13 | Core O motion; Ph group out-of-plane distortions | 8.1% | 1.1% | 437 | 18/19 | 14 | Core O motion; Ph group out-of-plane distortions | 7.3% | 2.6% |
| ca. 480 | 15 | 9 | Core O motion; coupled to C–I scissoring modes; Ph group out-of-plane distortions | 11.2% | 27.9% | ||||||
| 507 | 16 | 6 | Ph group out-of-plane distortions | 1.4% | 1.9% | 507 | 24 | 6 | Ph group out-of-plane distortions | 1.3% | 0.1% |
| 513 | 17 | 6 | Ph group out-of-plane distortions | 0.7% | 0.5% | 515 | 25 | 6 | Ph group out-of-plane distortions | 0.7% | 0.1% |
| 531 | 527 | ||||||||||
| 544 | 18 | 6 | Ph group out-of-plane distortions | 3.6% | 0.8% | 544 | 26 | 6 | Ph group out-of-plane distortions | 3.4% | 0.0% |
| 548 | 549 | ||||||||||
| 618 | 19 | 18 | Ph group in-plane distortions | 0.1% | 0.1% | 619 | 27 | 18 | Ph group in-plane distortions | 0.1% | 0.0% |
| ca. 675 | 20 | 6 | Ph group in-plane distortions | 0.1% | 0.2% | d | 28 | 6 | Ph group in-plane distortions | 0.1% | 0.0% |
| 691 | 21 | 32 | Ph group distortions; coupled to some C–I motions | 0.0% | 4.4% | d | 29 | 30 | Ph group distortions | 0.0% | 0.0% |
| 755 | 22 | 18 | Ph group distortions | 0.0% | 0.1% | d | 30 | 18 | Ph group distortions | 0.0% | 0.0% |
For Fig. 1 and 2, the theoretically predicted infrared spectra display myriad peaks throughout the 50–800 cm−1 region. All peaks predicted within the spectrum are numbered sequentially, starting at low energy, for both [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 (Fig. 1) and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 (Fig. 2). This peak numbering scheme is the same as that used in Table 1 for the assignments of peaks to predicted vibrational modes. Also shown in Fig. 1 and 2 are the experimental infrared spectra for [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 (represented as the upper solid lines in Fig. 1 and 2, respectively), at varying dilutions by mass with polyethylene (vide supra).
50–400 cm−1 region
The experimental spectra of the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 cluster in the 50–150 cm−1 region (Fig. 1a) contains two main features of interest. Below 100 cm−1, there is a broad, featureless peak with a maximum intensity at 65 cm−1, which we have previously attributed to phenyl group motions that are dampened in the solid phase compared with the gas phase calculation.15 The second feature, with maximum intensity at 107 cm−1, is assigned to peak #4 in the predicted spectrum. This peak arises from a combination of thirteen vibrational motions of varying infrared activity involving motions of the Au core. Based upon our previous work,15,16 this peak is expected to exhibit strongly within the experimental spectrum due the high Au core contribution component of 10.6%. The experimental spectrum of the [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 cluster below 150 cm−1 (Fig. 2a) is obscured by a moderate amount of fringing. These fringing effects are similar to that observed in our previous work for H4Ru4(CO)12,16 and arise due to multiple reflections between parallel reflective surfaces within the experimental setup; efforts to remove them were not successful.Between 150 and 300 cm−1, the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 spectrum is almost identical to the [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 spectrum with five well resolved peaks in the experimental spectrum appearing in the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 spectrum (and the [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 spectrum) at 187 (188), 209 (208), 227 (227), 255 (255), and 273 (274) cm−1. The calculated peak at 187 (188) cm−1 is assigned to peak #6 (#10) in the experimental spectrum, arises due to several Au–P stretches that have a high (>10%) contribution from the Au core. The features observed at 209 and 227 (208 and 227) cm−1 are tentatively assigned to peaks #7 and low-energy shoulders (#11/12), and #8 (#13), respectively, from the predicted spectrum. These predicted peaks correspond to numerous rocking motions of the phenyl groups, with little (<3%) contribution from either the core or the counter-ions. The experimental peaks at 255 and 273 (255 and 274) cm−1 are assigned to peaks #9 and #10 (#14 and #15) in the predicted spectra, which both arise due to Ph–P–Ph bending modes.
The experimental spectrum of the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 cluster within the 300–400 cm−1 region possesses a single large peak occurring at 330 cm−1. This peak is assigned to peak #11 in the predicted spectrum of [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2, which is due to two vibrational modes of each of the two BF−4 anions. These four vibrational modes, which are predicted to occur at 322, 323, 328, and 329 cm−1, are due to bending and scissoring vibrations of the two BF−4 counter-ions. These features are not theoretically predicted for the counter-ion alone in the gas phase, and were only found to appear when counter-ions were explicitly included in the calculation with [{{Au(PPh3)}3(μ3−O)}2]2+. The corresponding [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 experimental spectrum exhibits no corresponding MnO−4 experimental feature, though the peak is theoretically predicted to appear at ca. 370 cm−1 (peak #16), and is likely to be masked by the much more intense peak at 420 cm−1.
Fig. 3a compares the three predicted spectra for the 50–400 cm−1 region, for [{{Au(PPh3)}3(μ3−O)}2]2+, [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2, and [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2. Below 150 cm−1, the three predicted spectra differ markedly, with the [{{Au(PPh3)}3(μ3−O)}2]2+ predicted spectrum having much smaller intensities within this region, while [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 show differing intensities around 125–140 cm−1. Between 125 and 150 cm−1 there is expected to be considerable contribution to the vibrations due to the counter-ions in both the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 cases, which gives rise to these intensity differences, although these differences appear dampened in the experimental spectrum. Between 150 and 300 cm−1, there is general similarity between the three theoretical spectra, in both intensities and peak locations, while between 300 and 400 cm−1, a peak is predicted for both [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 (330 cm−1) and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 (370 cm−1) which is absent in the spectrum calculated for [{{Au(PPh3)}3(μ3−O)}2]2+ without counter-ions (vide supra).
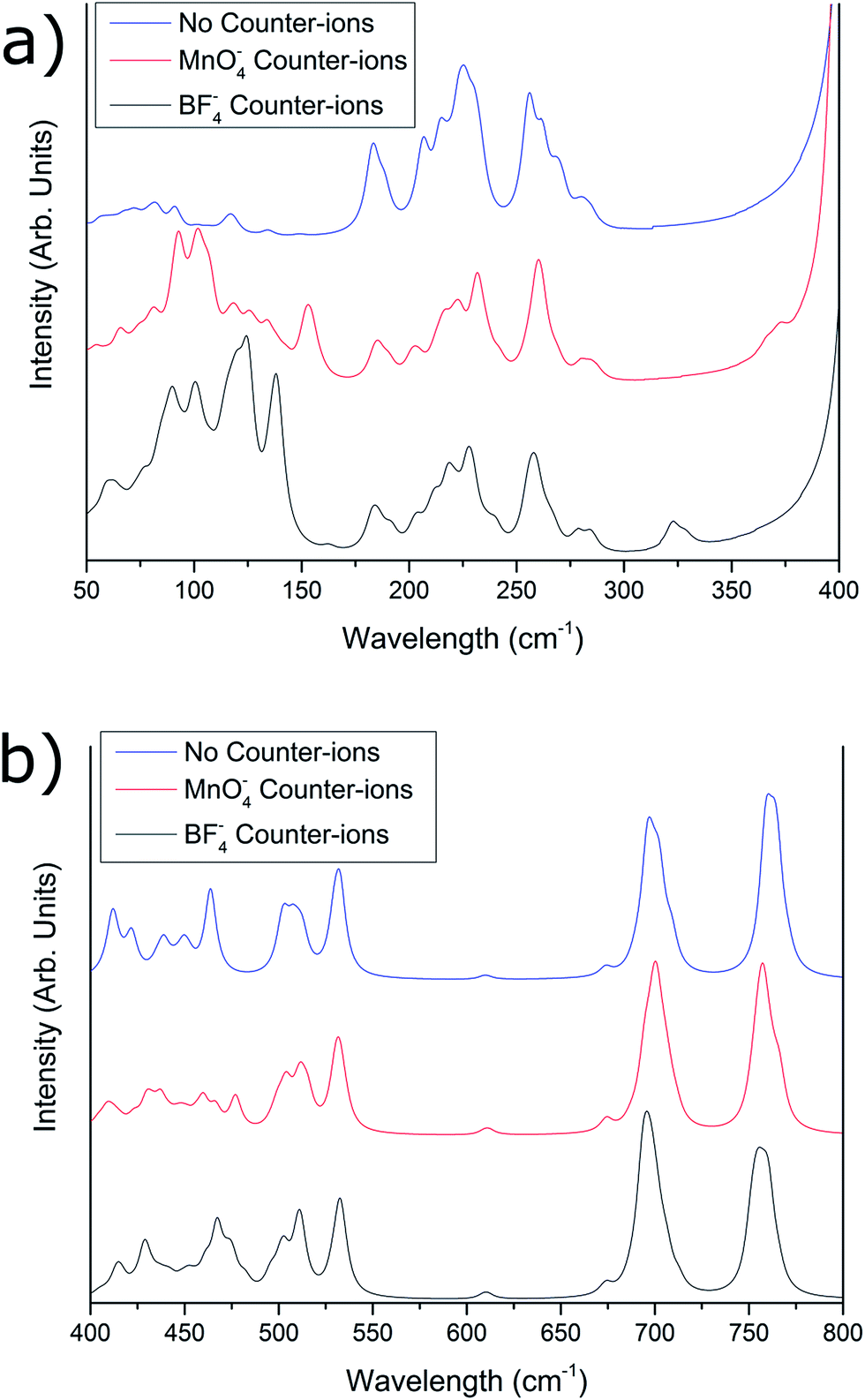 | ||
| Fig. 3 Comparison between theoretically calculated far-IR spectra of the tris(triphenylphosphinegold)oxonium salts with no counter-ions present (blue), MnO−4 counter-ions present (red), and BF−4 counter-ions present (black), in the (a) 50–400 cm−1 region and (b) 400–800 cm−1 region. The simulated spectra have been corrected by eqn (1). | ||
400–800 cm−1 region
The experimental spectra of [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 within the 400–800 cm−1 region are shown in Fig. 1b and 2b, respectively. Both spectra exhibit baseline between 700–750 cm−1 due to the complete absorption of the polyethylene used as the diluting agent in this region.26,27 The experimental spectrum between 400 and 500 cm−1 for [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 (and for the [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 spectrum) exhibits peaks at 420 (420) cm−1, 438 (439) cm−1, and a shoulder at ca. 480 (ca. 480) cm−1. For the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 spectrum, these peaks are assigned to theoretical peaks #12, #13, and #15, respectively, which are due to Ph group out-of-plane distortions, as well as a large contribution from core O motion (up to 11% for peak #15). Theoretical peak #15 is also predicted to possess a large component of counter-ion motion, almost 28% of the total atomic motion. The [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 peaks at 420 and 437 cm−1 are assigned to peaks #17 and #18/#19, respectively, which arise from Ph group out-of-plane distortions and core O atomic motion, similar to the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 spectrum. The former of these peaks is predicted to possess a significant 16% contribution of atomic motion from the counter-ions. The shoulder at ca. 480 cm−1 in the experimental [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 spectrum cannot be unambiguously identified, so is left unassigned.The spectrum between 500 and 600 cm−1 for [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 (and for the [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 spectrum) shows two doublets at 507 & 513 (507 & 515) cm−1, and 544 & 548 (544 & 549) cm−1, with a smaller intensity peak between them at 531 (527) cm−1. The former of the doublets is assigned to peaks #16 and #17 (#24 and #25) in the theoretical spectrum, while the higher energy of the doublets are assigned to peak #18 (#26). These peaks are predicted to arise due to eighteen moderate intensity (<100 km mol−1) phenyl group out-of-plane distortions, with little contribution (<4%) from the Au core or the counter-ions. The peak which is observed at 531 (527) cm−1 is unassigned to any peaks within the theoretical spectrum, and is of unknown origin.
Above 600 cm−1, three features are observed in the spectrum of [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2, at 618 cm−1, 691 cm−1, and 755 cm−1, as well as a shoulder at ca. 675 cm−1. The low intensity feature at 618 cm−1 matches a similar feature in the [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 spectrum at 619 cm−1, and are assigned to peaks #19 and #27, respectively, within the theoretical spectra. These peaks are predicted to arise due to eighteen very low infrared intensity (<5 km mol−1) vibrational modes, which are due to Ph group in-plane distortions.
The [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 peaks observed at 691 and 755 cm−1 are assigned to peaks #21 and #22 in the theoretical spectrum, and are predicted to arise due to Ph group distortions, with very little contribution (<0.1%) due to either the Au core or the counter-ions (<5%). The corresponding [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 features display significant noise due to the polyethylene absorption window within this region, however similar peaks to the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 spectrum are predicted (peaks #29 and #30), and are found to arise similarly due to Ph group distortions. Finally, a shoulder observed at ca. 675 cm−1 within the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 spectrum is assigned to predicted peak #20, also due to Ph group distortions.
The predicted spectra for [{{Au(PPh3)}3(μ3−O)}2]2+, [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2, and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 in the 400–800 cm−1 region are compared in Fig. 3b. Differences in relative intensities are observed for the 400–500 cm−1 region, which is attributed to differences in the counter-ion contribution toward the intensity of the peaks within this region, as discussed above. Above 500 cm−1, no significant differences within the spectra were predicted to occur. Experimentally, almost no differences were observed between the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 spectra, outside of the 330 cm−1 peak present in the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 spectrum. This suggests a dampening of all counter-ion vibrational modes when the sample is in the solid phase, removing the effect of these modes on the infrared spectrum.
Binding energies
The dimerization of the [{{Au(PPh3)}3(μ3−O)}2]2+ species involves the formation of two Au–Au bonds across one edge of the Au3 trimer. The two monomers are bound centrosymmetrically, with the six gold atoms forming a ring in a chair conformation (see Fig. S1–S3; ESI†). Within the monomer, the triphenylphosphine ligands are positioned such that an edge of the Au3 moity is exposed, facilitating the dimerization of the species with minimal ligand repositioning, hence very little bond strain is anticipated to accompany this. The energy of dimerization of the [{{Au(PPh3)}3(μ3−O)}2]2+ system is calculated to be 1.10 eV when no counter-ions are present. With the addition of counter-ions, the energy increases markedly; for the dimer [{{Au(PPh3)}3(μ3−O)}2]2+ system with an MnO−4 counter-ion, the energy of dimerization is predicted to be 3.20 eV, and 3.06 eV for the BF−4 counter-ion. This significant increase in dimerization energy can be attributed to the electrostatic repulsion between the monomers when no counter-ion is present, resulting in a much less favourable dimerization interaction.For both the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 systems, the binding energy of the respective counter-ions was calculated by the stepwise removal of the two counter-ions. For [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2, this was determined to be 3.06 eV and 4.65 eV for the removal of the first and second counter-ion, respectively. For [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2, this is slightly higher at 3.15 eV and 4.74 eV, respectively. Where the systems are not dimerized, the removal of BF−4 from [{{Au(PPh3)}3(μ3−O)}2]2+ is calculated at a lower 2.87 eV, while the removal of MnO−4 is predicted to be 2.90 eV.
The far-infrared vibrational spectrum of the monomer species differs from that of the dimers (see Fig. S4; ESI†). In particular, large differences are predicted between 400 and 475 cm−1, with several features absent from the monomer spectrum that are present in the dimer spectrum. As well, above 475 cm−1 all features are expected to display significant differences in relative intensities, between the monomer and dimer spectra.
Conclusions
The far-infrared spectrum of [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 is found to possess a large feature at 330 cm−1, attributable to several counter-ion vibrational modes, which are only correctly predicted upon explicit inclusion of counter-ions within the calculations. A feature around 107 cm−1 was observed in the [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 infrared spectrum, which was assigned to 21 distinct vibrational modes arising due to Au–Au bond stretching, and motion of the Au core. Within the 400–800 cm−1 region, little dependence of the theoretical spectra on the nature of the counter-ions was predicted, while in the 50–400 cm−1 region, the relative intensities of predicted vibrational modes was found to depend heavily on the presence and nature of the counter-ions. Finally, the dimerization energies of both [{{Au(PPh3)}3(μ3−O)}2]2+(BF−4)2 and [{{Au(PPh3)}3(μ3−O)}2]2+(MnO−4)2 were calculated, and found to be 3.06 eV and 3.20 eV, respectively, when the counter-ions were explicitly included within the calculation, and only 1.10 eV when the counter-ions were not included.Acknowledgements
This research was undertaken on the High-Resolution Infrared beamline at the Australian Synchrotron, Victoria, Australia. Financial support from the Centre for Energy Technologies at the University of Adelaide, the MacDiarmid Institute for Advanced Materials and Nanotechnology, the University of Canterbury and the University of Malaya is gratefully acknowledged. Computing resources provided by the National Computational Infrastructure (NCI) Facility and eResearch SA is also gratefully acknowledged. The authors would also like to thank Dr Dominique Appadoo for his advice on recording the infra-red spectra at the Australian Synchrotron.References
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- A. Nesmeyanov, E. Perevalova, D. Lemenovskii, V. Dyadchenko and K. Grandberg, Russ. Chem. Bull., 1974, 23, 1587–1589 CrossRef.
- A. Nesmeyanov, E. Perevalova, Y. T. Struchkov, M. Y. Antipin, K. Grandberg and V. Dyadhenko, J. Organomet. Chem., 1980, 201, 343–349 CrossRef CAS.
- E. Smyslova, E. Perevalova, V. Dyadchenko, K. Grandberg, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1981, 215, 269–279 CrossRef CAS.
- E. Perevalova, K. Grandberg, V. Dyadchenko and T. Baukova, J. Organomet. Chem., 1981, 217, 403–413 CrossRef CAS.
- E. Perevalova, T. Baukova, M. Sazonenko and K. Grandberg, Russ. Chem. Bull., 1985, 34, 1722–1726 CrossRef.
- K. Grandberg and V. Dyadchenko, J. Organomet. Chem., 1994, 474, 1–21 CrossRef CAS.
- T. Baukova, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1981, 220, 125–137 CrossRef CAS.
- T. Baukova, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1981, 221, 375–384 CrossRef CAS.
- A. Peregudov, L. Usatova, E. Smyslova and D. Kravtsov, Russ. Chem. Bull., 1994, 43, 125–129 CrossRef.
- H. Schmidbaur, A. Grohmann, M. E. Olmos and A. Schier, Patai's Chemistry of Functional Groups, 1999 Search PubMed.
- B. Djordjevic, K. A. Porter, S. Nogai, A. Schier and H. Schmidbaur, Organometallics, 2003, 22, 5336–5344 CrossRef CAS.
- M. I. Bruce and B. K. Nicholson, J. Chem. Soc., Chem. Commun., 1982, 1141–1143 RSC.
- M. I. Bruce and B. K. Nicholson, Organometallics, 1984, 3, 101–108 CrossRef CAS.
- J. F. Alvino, T. Bennett, D. P. Anderson, B. Donoeva, D. Ovoshchnikov, R. Adnan, D. R. Appadoo, V. Golovko, G. G. Andersson and G. F. Metha, RSC Adv., 2013, 3, 22140–22149 RSC.
- T. Bennett, R. H. Adnan, J. F. Alvino, V. Golovko, G. G. Andersson and G. F. Metha, Inorg. Chem., 2014, 53, 4340–4349 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- T. Dunning Jr and P. Hay, Mod. Theor. Chem., 1977, 3, 1–28 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS PubMed.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; B. Mennucci; G. A. Petersson; H. Nakatsuji; M. Caricato; X. Li; H. P. Hratchian; A. F. Izmaylov; J. Bloino; G. Zheng; J. L. Sonnenberg; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; J. J. A. Montgomery; J. E. Peralta; F. Ogliaro; M. Bearpark; J. J. Heyd; E. Brothers; K. N. Kudin; V. N. Staroverov; R. Kobayashi; J. Normand; K. Raghavachari; A. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; N. Rega; N. J. Millam; M. Klene; J. E. Knox; J. B. Cross; V. Bakken; C. Adamo; J. Jaramillo; R. Gomperts; R. E. Stratmann; O. Yazyev; A. J. Austin; R. Cammi; C. Pomelli; J. W. Ochterski; R. L. Martin; K. Morokuma; V. G. Zakrzewski; G. A. Voth; P. Salvador; J. J. Dannenberg; S. Dapprich; A. D. Daniels; Ö. Farkas; J. B. Foresman; J. V. Ortiz; J. Cioslowski and D. J. FoxGaussian 09 (Revision D.01), Gaussian 09 (Revision D.01), Gaussian, Inc.: Wallingford CT, 2009 Search PubMed.
- L. Kuz'mina, A. Churakov, K. Grandberg and J. Howard, J. Chem. Crystallogr., 2005, 35, 635–640 CrossRef.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- L. Lin, P. Claes, P. Gruene, G. Meijer, A. Fielicke, M. T. Nguyen and P. Lievens, ChemPhysChem, 2010, 11, 1932–1943 CAS.
- S. Krimm, C. Liang and G. Sutherland, J. Chem. Phys., 1956, 25, 549–562 CrossRef CAS PubMed.
- H. Thompson and P. Torkington, Trans. Faraday Soc., 1945, 41, 246b–260 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Containing a comparison between the crystallographic and DFT-optimized Au6O2 core, optimized structures for the [[(Ph3PAu)3O]BF4]2 and [[(Ph3PAu)3O]MnO4]2 species, and a comparison between the theoretical spectra for the [(PPh3Au)3O]− and ([(PPh3Au)3O]−)2 species. See DOI: 10.1039/c5ra11599j |
| This journal is © The Royal Society of Chemistry 2015 |