
Ionic liquids as buffer additives in ionic liquid-polyacrylamide gel electrophoresis separation of mixtures of low and high molecular weight proteins†
Abstract
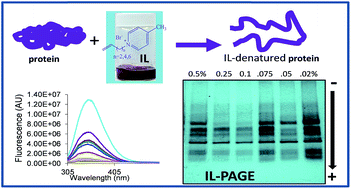
This study aims at investigating methodologies for better separation of proteins using novel hydrophobic ionic liquids (ILs). In this regard, hydrophobic ILs [CnPBr] (n = 4, 6, 8) were synthesized and examined in ionic liquid-polyacrylamide gel electrophoresis (IL-PAGE) as buffer additives for separation of catalase (Cat), transferrin (Tf), bovine serum albumin (BSA), ovalbumin (Ova) and α-lactalbumin (α-Lact). The influence of alkyl chain length of the cation of these ILs and their concentration in running and sample buffers on protein separation was investigated. Separation using ILs as additives was achieved at lower concentrations as compared to standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The IL concentrations were 100-fold less in sample buffer and 5-fold less in running buffer as compared to conventional SDS-PAGE. The results demonstrated that ILs additives played a role in improving some protein separation, IL-PAGE provided higher resolution and separation efficiency than SDS-PAGE for Tf and Ova. Fluorescence studies were performed in order to understand protein–IL interactions and were used to determine the appropriate IL for use as a buffer additive in PAGE. When compared with standard SDS-PAGE, no heating of sample buffer was required in IL-PAGE, which revealed that proteins could be efficiently denatured by use of IL, which was later confirmed by use of circular dichroism (CD) studies.
 Please wait while we load your content...
Please wait while we load your content...

