
A better understanding of the capacity fading mechanisms of Li3V2(PO4)3†
Liping Wang*a,
Jin Xua,
Chong Wanga,
Xumei Cuiab,
Jingze Lia and
Yong-Ning Zhou*c
aState Key Laboratory of Electronic Thin Films and Integrated Devices, University of Electronic Science and Technology of China, Chengdu 610054, China. E-mail: lipingwang@uestc.edu.cn; ynzhou@fudan.edu.cn; Fax: +86 28 83202569; Tel: +86 28 83207620
bSichuan Key Lab of Comprehensive Utilization of Vanadium and Titanium Resources, Panzhihua University, Panzhihua 617000, China
cDepartment of Materials Science, Fudan University, Shanghai, 200433, China
First published on 18th August 2015
Abstract
Lithium vanadium phosphate Li3V2(PO4)3 (LVP) has emerged as an appealing cathode material for next generation lithium-ion batteries owing to its high theoretical capacity (197 mA h g−1) and high average working potential around 4.0 V vs. Li/Li+. However, the capacity fading problem limits its practical application, especially when a high cut-off voltage over 4.3 V is applied. In this study, the capacity fading mechanisms of LVP in different voltage windows of 3.0–4.3 V and 3.0–4.8 V are studied systematically by using electrochemical impedance spectroscopy, a galvanostatic intermittent titration technique, cyclic voltammetry, and in situ X-ray absorption spectroscopy. Surprisingly, the structure of LVP can be fully recovered after one cycle (even for a cut-off voltage as high as 4.8 V). It indicates that LVP is a promising cathode system with excellent structure reversibility intrinsically. We revealed that the capacity fading during high voltage cycling is mainly due to the parasitic reaction with the electrolyte, kinetics limitation and V dissolution, rather than LVP structure degradation. In addition, a “crystalline–amorphous–crystalline” phase transition pathway was revealed during the LVP synthesis process of the solid-state reaction by using synchrotron based in situ X-ray diffraction.
1. Introduction
Next generation lithium ion batteries require electrode materials with high energy density, high power density and long cycle life.1 Lithium metal phosphates contain both mobile Li cations and redox-active metal ions shrouded within a rigid phosphate network, offering advantages of safety, cost effectiveness and good environmental compatibility, which make them promising electrode candidates for applications in electric and hybrid vehicles.2 Among them, lithium vanadium phosphate (Li3V2(PO4)3, denoted as LVP) with a NASICON structure delivers a theoretical capacity of 197 mA h g−1 as three lithium ions are intercalated/deintercalated per formula unit with the average operational voltage at 4.0 V vs. Li/Li+, guaranteeing a high energy density and fast lithium-ion diffusion.3–5 It also plays a vital role in improving the electrochemical performance of Li3V2(PO4)3/LiFePO4 composite electrodes at room temperature and low temperature.6–8Similar as olivine-type LiMPO4 (M = Fe, Mn etc.), LVP also suffers from low electronic conductivity (2 × 10−8 S cm−1).9 Minimizing particle sizes,10–12 coating with conductive layers13–15 and doping with other transition metals16–18 are common approaches to circumvent this issue, which makes the cycling performance and rate capability comparatively improved. However, good capacity retention can only be obtained when two lithium ions in Li3V2(PO4)3 are extracted and inserted with a capacity of 133 mA h g−1 in the voltage window 3.0–4.3 V. In situ X-ray diffraction and X-ray absorption spectroscopy in the voltage window 3.0–4.3 V demonstrated that the crystal and electronic structure could be fully recovered when two lithium ions were extracted and inserted.12,19 If the third lithium is extracted from LVP when charging to a higher voltage (over 4.5 V), its capacity fading will be quite significant,12 so it is very difficult to utilize the full theoretical capacity of 197 mA h g−1 for LVP. Huang et al. proposed that the capacity fading was mainly due to the oxidation of electrolyte at high voltage.4 Yin et al. thought it could be the result of poor kinetics when the third Li ion was extracted.9 Kang et al. performed the in situ X-ray diffraction (XRD) to monitor the crystal structure change of LVP during charge and discharge in the voltage window 3.0–4.8 V.12 They found the major diffraction peaks of LVP can be fully recovered after one cycle even with capacity fading, indicating an unexpected structural reversibility. The only difference is a small increase in the unit cell dimensions of LiV2(PO4)3 formed during discharging. To the best of our knowledge, the mechanism of the capacity fading for LVP in a large voltage window 3.0–4.8 V is still not well understood.
In this work, the capacity fading mechanism of LVP in the voltage range of 3.0–4.8 V was investigated systematically by using electrochemical tests including electrochemical impedance spectroscopy (EIS), galvanostatic intermittent titration technique (GITT), studying the phase stability at room temperature via X-ray diffraction as well as the electronic structure stability in situ X-ray absorption spectroscopy (XAS). In addition, the phase transition pathway during LVP synthesis process of solid-state reaction was studied by in situ X-ray diffraction for the first time.
2. Experimental and characterizations
2.1 Material synthesis
Li3V2(PO4)3 was prepared via a conventional solid state reaction route. All the chemicals are of analytical grade. The mixture of stoichiometric amounts Li2CO3 (0.015 mol, 1.108 g), NH4VO3 (2.339 g), NH4H2PO4 (3.451 g) and citric acid (1.710 g) were used as starting materials. After a high energy ball-milling process at 300 rounds per minute for 12 hours, the mixture was annealed at 700 °C for 10 hours under Ar/H2 atmosphere at a heating ramp of 5 °C min−1.2.2 Electrochemical tests
The electrode was prepared by mixing active LVP/C composite (80 wt%), carbon black (10 wt%), and polyvinylidene fluoride (PVDF, 10 wt%) in N-methylpyrrolidone (NMP) solvent to form a homogenous slurry. The slurry was then coated onto an Al foil and dried at 100 °C for 12 h in a vacuum oven to form the electrodes. After that, the electrodes were incorporated into cells with a high-purity lithium metal foil anode and a Celgard separator. Commercial electrolyte LP30 (1 M LiPF6 in a mixture of ethylene carbonate (EC), dimethyl carbonate (DMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume)) was used as the electrolyte. The 2032-type coin cells were assembled in an argon-filled glove box for electrochemical tests. The galvanostatic charge–discharge test and galvanostatic intermittent titration technique (GITT) were carried out on a LAND CT2001A battery test system. For the GITT, the cells were charged at a current density of 13.3 mA g−1 for 15 minutes, followed by open circuit relaxation for 4 hours. The cyclic voltammetry curves were measured using a CHI66C electrochemical working station at different scanning rates. Electrochemical impedance spectroscopy (EIS) measurements were performed on cells at different charge–discharge status using an electrochemical workstation (VMP multipotentiostat). The amplitude of the a.c. signal was 10 mV over the frequency range of 10−2 and 106 Hz. All the electrochemical measurements were carried out at room temperature. The weight of the active material is about 2.0 mg cm−2.
:1 by volume)) was used as the electrolyte. The 2032-type coin cells were assembled in an argon-filled glove box for electrochemical tests. The galvanostatic charge–discharge test and galvanostatic intermittent titration technique (GITT) were carried out on a LAND CT2001A battery test system. For the GITT, the cells were charged at a current density of 13.3 mA g−1 for 15 minutes, followed by open circuit relaxation for 4 hours. The cyclic voltammetry curves were measured using a CHI66C electrochemical working station at different scanning rates. Electrochemical impedance spectroscopy (EIS) measurements were performed on cells at different charge–discharge status using an electrochemical workstation (VMP multipotentiostat). The amplitude of the a.c. signal was 10 mV over the frequency range of 10−2 and 106 Hz. All the electrochemical measurements were carried out at room temperature. The weight of the active material is about 2.0 mg cm−2.
2.3 Ex situ and in situ structural characterizations
Crystal structures of the samples were determined by X-ray diffraction (XRD) (λ = 0.7787 Å) at beamline X14A of National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory. A silicon strip detector was mounted on the 2θ arm of a 6-circle Huber diffractometer, about 1400 mm from the sample. The detector has 640 channels, each of which covers 0.005° in 2θ. The XRD patterns were taken by scanning the 2θ arm at 2° per step. Each scan took about 6 minutes with a 2θ angular range from 4° to 50°. The lattice parameters of the as-obtained samples were obtained from Rietveld refinement method using Fullprof software.20 In situ XRD measurement during heating (from room temperature to 700 °C) was carried out to examine the phase transition during the synthesis process in the real time. The mixture of precursors after ball-milling was sealed into quartz tube in the Ar-filled glove box and heated by heating coil during XRD measurement. In situ V K-edge XAS data were collected in transmission mode at beamline X18A of NSLS. Energy calibration was carried out using the first inflection point of the reference spectrum of V metal foil (V K-edge = 5465 eV), which was simultaneously collected during each measurement during the in situ experiments. The absorption spectra were measured within the interval [−100 eV to 650 eV] relative to the V K-edge. The X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectrum were processed using the Athena18 and Artemis19 software packages.21 The AUTOBK code was used to normalize the absorption coefficient, and to separate the EXAFS signal, χ(k), from the isolated atom-absorption background. The extracted EXAFS signal, χ(k), was weighted by k3 to emphasize the high-energy oscillations and then Fourier-transformed in a k range from 3.2 to 11.5 Å−1 using a Hanning window with a window sill (Δk) of 1.0 Å−1, thereby obtaining magnitude plots of the EXAFS spectra in R-space (Å). The Fourier-transformed peaks were not phase-corrected, and thus the actual bond lengths are approximately 0.3–0.5 Å longer. The morphology was characterized on a field-emission scanning electron microscope (FE-SEM, Hitachi, S3400N) and the compositions were analyzed with an energy dispersive X-ray spectroscopy (EDS, Oxford INCA PentaFET-x3).3. Results and discussion
Understanding phase transition pathway during material synthesis process is very important for controlling and optimizing the synthesis procedures in order to obtain the ideal final product. Benefit from the high flux of synchrotron radiation, synchrotron-based in situ X-ray diffraction provides a powerful method for direct observation of phase transition process during reaction in real time.22 To monitor the phase transition pathway of the LVP synthesis process via solid state reaction, synchrotron based in situ X-ray diffraction during heating was utilized. The setup illustration for the in situ experiment is shown in Fig. 1a. The time-resolved XRD patterns from ball-milled precursor mixture to the final LVP phase were recorded from room temperature (RT) to 700 °C under Ar atmosphere, as shown in Fig. 1b. At the beginning of heating process, a group of broad peaks are observed, which can be assigned to Li2CO3 (JCPDS no. 83-1454), NH4VO3 (JCPDS no. 09-0411) and NH4H2PO4 (JCPDS no. 78-2415). The peak broadening indicates that ball-milling process can decrease the particle size of the precursors effectively and result in a quite homogeneous mixture, which are very important for obtaining high quality products. From RT to 415 °C, all the diffraction peaks shift to lower 2θ angles along with the temperature increase, which is owing to the thermal expansion of the crystal lattice,23 but no remarkable phase transition is observed. Concomitantly, the peak intensity decreases and finally disappears at 415 °C, indicating that the precursors Li2CO3, NH4VO3, NH4H2PO4 decompose gradually into nano-cluster and/or amorphous state with temperature increase. From 415 °C to 630 °C, no clear peak can be observed, implying that all of the precursors decompose and react with each other in this temperature range. At the same time, the nucleation of the new phases may have happened, but they cannot be detected, because their size is smaller than the coherence length of X-ray. From 630 °C to 700 °C, new diffraction peaks emerge and grow up immediately. The new peaks can be well indexed by LVP phase (JCPDS no. 78-1106), demonstrating the formation of well crystallized LVP. This “crystalline–amorphous–crystalline” pathway during LVP synthesis process is quite similar as that in the synthesis process of LiFePO4 using FePO4 and Li2CO3 as precursors.24 Fig. 1c shows the powder XRD pattern of the as-obtained LVP at room temperature. Rietveld refinement of the pattern indicates a monoclinic structure with a space group of P21/n. The lattice parameters are a = 8.607 Å, b = 8.594 Å, c = 12.041 Å (β = 90.567). The SEM image of the as-obtained LVP shown in Fig. 1d illustrates that the particle size of this material is in micrometer dimension. The carbon content in the final product is about 6.3 wt% measured by thermogravimetry test as shown in Fig. S1.†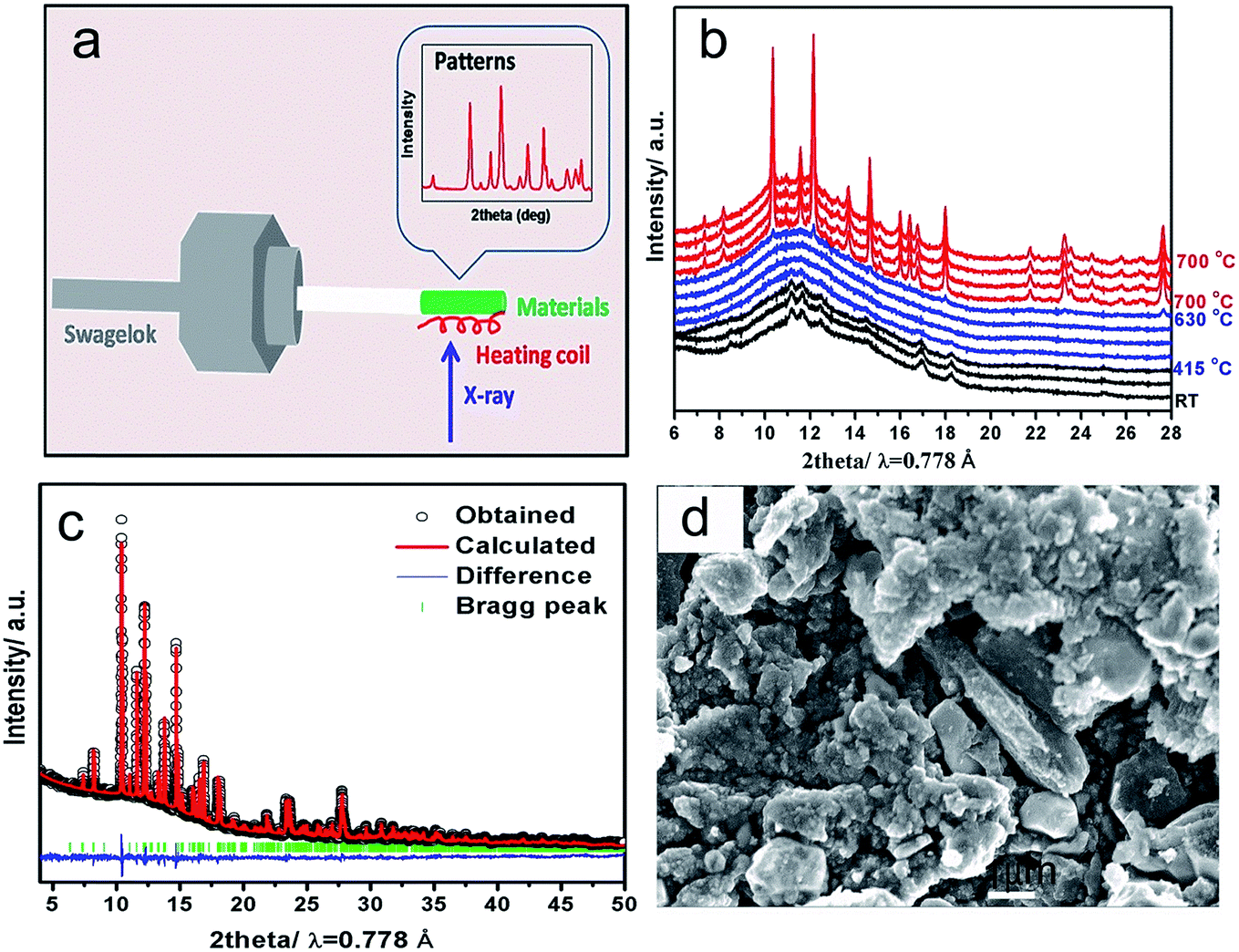 | ||
| Fig. 1 (a) Schematic illustration of the in situ solid-state reaction reactor; (b) time-resolved in situ XRD patterns during heating the precursors from RT to 700 °C at a ramp rate of 5 °C min−1; (c) Rietveld refinement of the XRD pattern for as-obtained LVP with observed data (black points), calculated data (red line), difference (blue line) and Bragg positions (green bars). Rwp = 5.56%, Rexp = 1.19%, Rp = 3.24%; (d) SEM image of the as-obtained LVP. | ||
The electrochemical performance of the as-obtained LVP was evaluated by galvanostatic charge–discharge test. The typical charge–discharge curves in different voltage windows (3.0–4.3 V and 3.0–4.8 V versus Li+/Li) are presented in Fig. 2a. During the charge process below 4.3 V, three distinct plateaus at 3.6 V, 3.7 V and 4.1 V can be observed. Based on the previous study,4 they correspond to three two-phase transition processes: Li3V2(PO4)3 → Li2.5V2(PO4)3, Li2.5V2(PO4)3 → Li2V2(PO4)3 and Li2V2(PO4)3 → Li1V2(PO4)3, respectively. When the cutoff voltage is set to 4.3 V, the discharge curve is quite symmetric with the charge curve, indicating excellent reversibility below 4.3 V. When the cell is charged to 4.8 V, a sloppy plateau can be observed around 4.5 V, which is attributed to the extraction of the last Li (Li1V2(PO4)3 → Li0V2(PO4)3).4 Interestingly, the discharge curve after charging to 4.8 V is quite different from that after charging to 4.3 V. It shows a large slope to 3.6 V rather than distinguished plateaus, which are more like a solid-solution behavior. Yin et al. attributed this “solid-solution region” to the disordered lithium insertion in V2(PO4)3, which has the mixed V4+/V5+ state with no charge ordering.9 The asymmetry of the charge–discharge process suggests that extraction of the third lithium (Li1V2(PO4)3 → Li0V2(PO4)3) can cause significant lattice changes, leading to different kinetic or thermodynamic process, which will be discussed in details later. The capacity retention performance in the first 40 cycles is presented in Fig. 2b. When this operation voltage window is restricted between 3.0 and 4.3 V (corresponding to two lithium ions in LVP are extracted and inserted) at the current rate of 13.3 mA g−1, an initial discharge capacity of 119 mA h g−1 can be obtained, which decreases to 115 mA h g−1 after 40 cycles with a capacity retention rate of 97%. However, with fully (de)lithiation between 3.0–4.8 V, the cell suffers from severe capacity fading from 158 mA h g−1 to 135 mA h g−1 with a capacity retention of only 85% after 40 cycles. The main capacity loss in the large electrochemical window 3.0–4.8 V should be highly related to the slope around 4.5 V during the third lithium extraction as well as a parasitic reaction with electrolyte in high voltage window. In order to obtain the working potential in the equilibrium condition, galvanostatic intermittent titration technique (GITT) was carried out with relaxation time of 4 hours in the voltage window 3.0–4.8 V as shown in Fig. 2c (Fig. S2† for 3.0–4.3 V) and its corresponding voltage polarization is shown in Fig. 2d. In the charge process below 4.2 V, the voltage hysteresis for the first two lithium extraction from LVP is less than 0.1 V, comparable to typical lithium transition metal phosphates.25 However, when charge over 4.2 V, the voltage hysteresis increases significantly up to 0.5 V during the third lithium extraction. After relaxation, a flat plateau can be observed at around 4.3 V, which implies that the slope between 4.3 V and 4.8 V in the charge–discharge curves is due to the kinetics limitation. Nazar et al.26 ascribed this high voltage polarization to the lower electronic/ionic conductivity of V2(PO4)3. It could also be owing to high phase boundary energy similar as transition metal oxide whose voltage hysteresis is even higher than 0.5 V.27 In the discharge process from 4.8 V to 3.7 V, the curves after relaxation still shows a slope rather than flat plateau, with very little voltage hysteresis, indicating that the solid solution behavior during discharge is not caused by kinetics limitation, but an intrinsic characteristics. It could be a disordered insertion process instead of phase transition.9
 | ||
| Fig. 2 (a) Typical galvanostatic charge–discharge curves (the 2nd cycle) of LVP in two different voltage windows: 3.0–4.3 V and 3.0–4.8 V; (b) cycling performance of LVP in voltage window 3.0–4.3 V and 3.0–4.8 V; (c) GITT curves plotted with the voltage as a function of specific capacity in the voltage window 3.0–4.8 V. The current density is set at 13 mA g−1 with a relaxation time of 4 hours; (d) the over-potential value as a function of specific capacity in the charge process. | ||
In order to better understand the capacity fading mechanism during charge–discharge process at the voltage window 3.0–4.8 V, electrochemical impedance spectroscopy (EIS) at various open circuit potentials were recorded and shown in Fig. 3. All of the spectra consist of a well-defined semicircle followed by a linear portion. Generally, the semicircle reflects the charge-transfer resistance (Rct) and interfacial capacitance between the electrolyte and the active material. The sloppy line in the low-frequency range represents the Warburg impedance, which is associated with lithium-ion diffusion in the LVP particles. An equivalent circuit (inset in Fig. 3b and d) is used to fit these impedance spectra using Zview software. Their corresponding charge transfer resistance is shown in Fig. 3b and d. In the voltage window 3.0–4.3 V with two lithium extraction/insertion, the Rct increases from the initial 33 Ω to 73 Ω after one cycle. In contrast, in the voltage window 3.0–4.8 V with three lithium extraction/insertion, a huge increase of Rct can be observed after charging to 4.8 V. The high resistance is kept during the following discharge process, which is about three times of that in the voltage window 3.0–4.3 V. The main increase comes from the third lithium extraction (Li1V2(PO4)3 → Li0V2(PO4)3) stage, which coincides with the high voltage hysteresis in our GITT measurements.
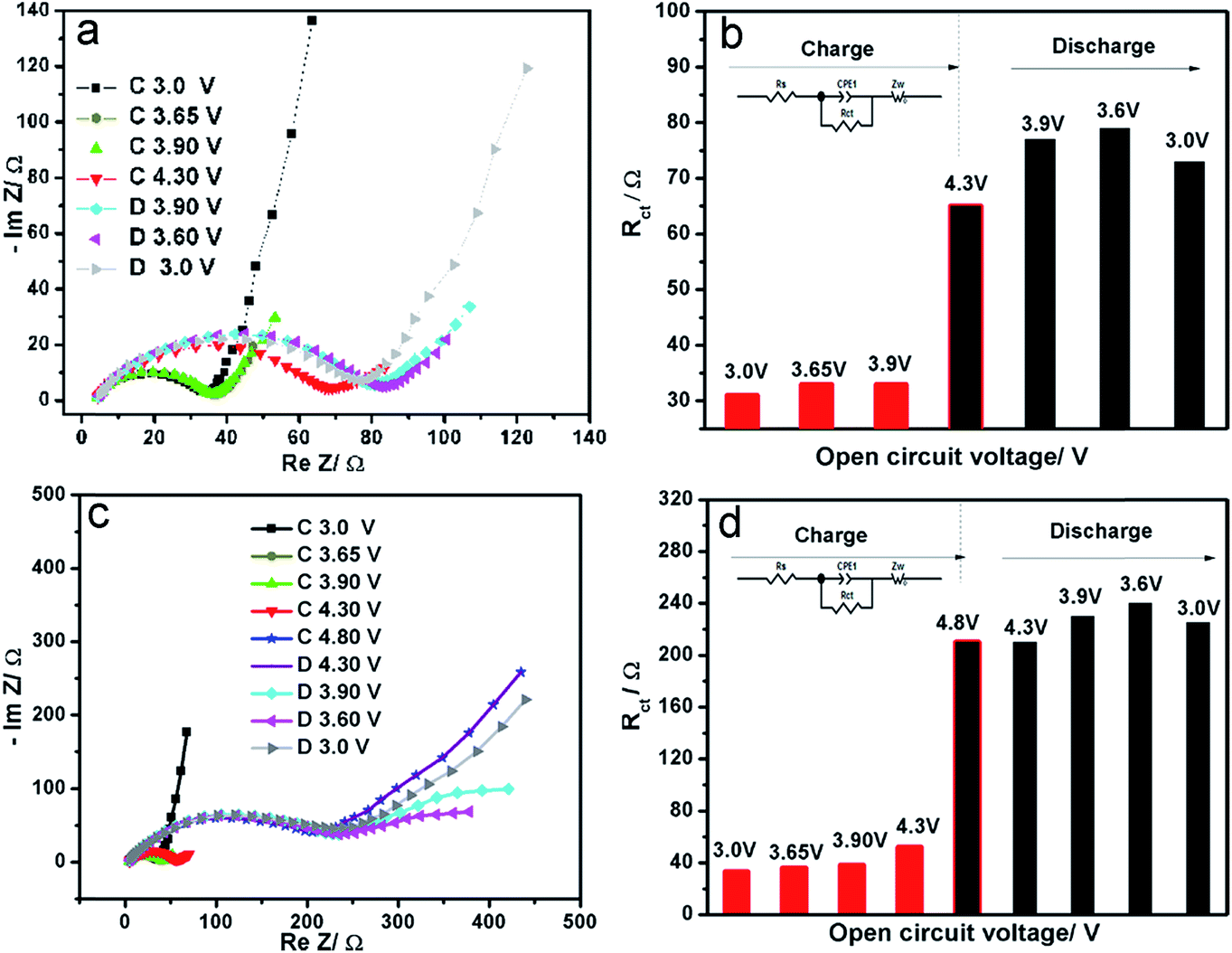 | ||
| Fig. 3 Electrochemical impedance spectroscopy (a) and corresponding charge-transfer resistance (b) of LVP at different charge–discharge states between 3.0–4.3 V in the initial cycle; electrochemical impedance spectroscopy (c) and corresponding charge-transfer resistance (d) at different charge–discharge states between 3.0–4.8 V in the initial cycle. | ||
The typical cyclic voltammetry curves of LVP in different voltage windows are shown in Fig. 4. The cathodic peaks are very symmetric with the anodic peaks in the voltage window 3.0–4.3 V, while they are quite asymmetric in the voltage window 3.0–4.8 V, indicating different kinetics process in the lithium extraction/insertion process (Fig. 4a–c). The peak current (Ip) increases with the increasing scan rates. The anodic peaks shift to higher potential and the cathodic peaks shift to lower potential with increasing scan rate. Meanwhile, the variation of peak current (Ip) is linear with the square root of scanning rate (v1/2), as shown in Fig. 4d, which implies a typical diffusion-controlled process. The chemical diffusion coefficient can be calculated from the Randles–Sevcik equation:
IP = 0.4463n3/2F3/2CLiSR−1/2T−1/2![[D with combining tilde]](https://www.rsc.org/images/entities/i_char_0044_0303.gif) Li1/2ν1/2 Li1/2ν1/2
| (1) |
485.4 C mol−1), C is the Li-ion concentration in Li3V2(PO4)3 (0.022 mol cm−3, 12 lithium ions per unit cell with a volume of 890.727 A3), S is the surface area of the electrode (cm2), R is the gas constant (8.314 J mol−1 K−1), T is the absolute temperature (K), CV is the chemical diffusion coefficient (cm2 s−1), v is the scanning rate (V s−1). Based on eqn (1), the apparent diffusion coefficients of Li-ion are 1.1 × 10−8 cm2 s−1 (3.0–4.3 V) and 9.0 × 10−9 cm2 s−1 (3.0–4.8 V), which are similar with the results obtained by EIS and GITT techniques.28 It reveals that charging to a higher voltage leads to a lower chemical diffusion coefficient, which could be related to the structural evolution and surface reaction. The chemical diffusion coefficient is much higher than that of LiFePO4 (10−14 cm2 s−1),29 suggesting that Li3V2(PO4)3 could be a good candidate as a high power electrode material.
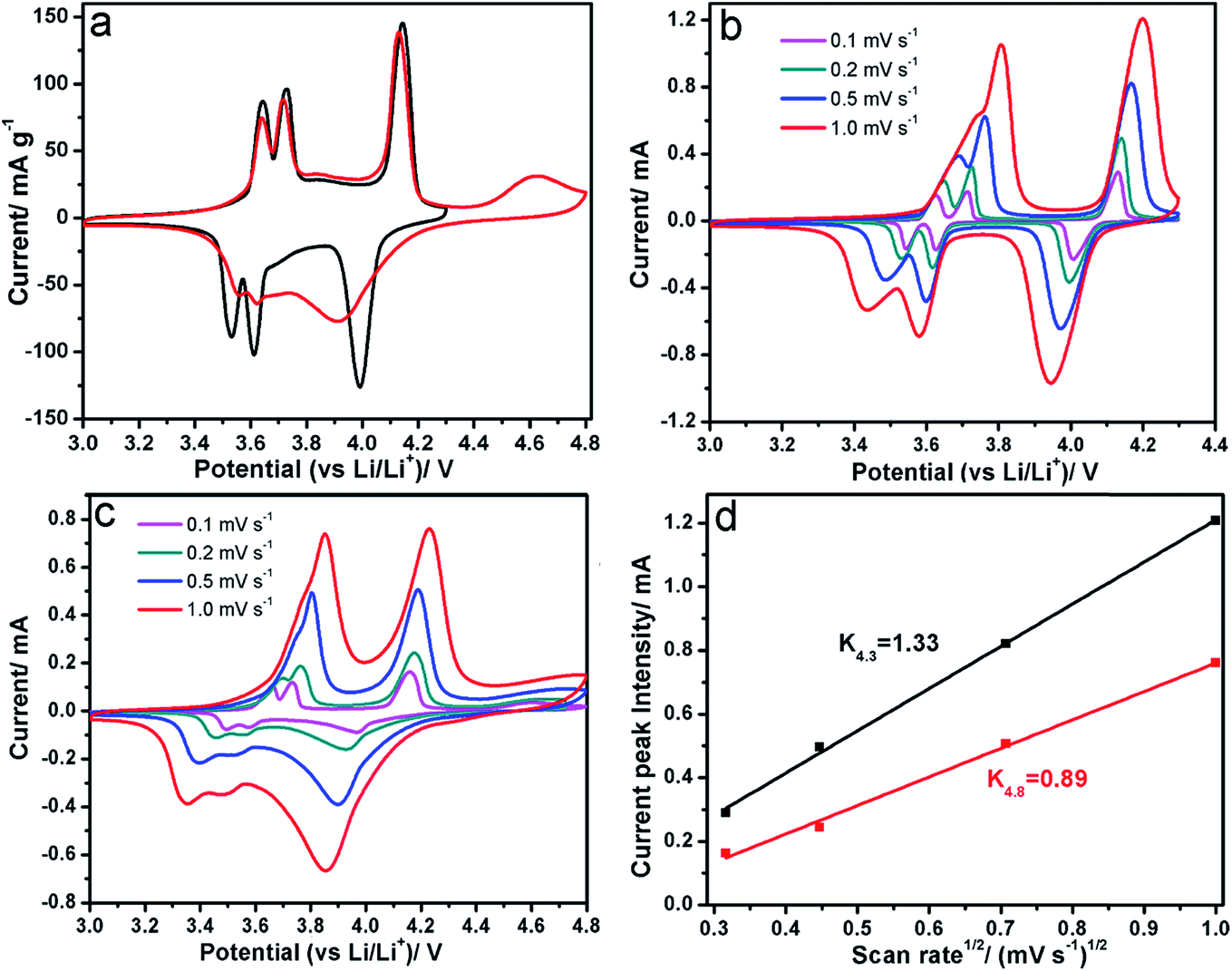 | ||
| Fig. 4 (a) Cyclic voltammetry curves of as-obtained LVP in voltage windows of 3.0–4.3 V (black) and 3.0–4.8 V (red) at a scan rate of 0.1 mV s−1; (b) cyclic voltammetry curves at different scan rates in 3.0–4.3 V; (c) cyclic voltammetry curves at different scan rates in 3.0–4.8 V; (d) peak current against square root of scan rate in different voltage windows based on the Li2V2(PO4)3 → Li1V2(PO4)3 oxidation peak. | ||
In situ X-ray absorption spectroscopy (XAS) at the V K-edge was carried out to monitor the valence state and local structure changes of V in LVP during charge and discharge (Fig. 5). Fig. 5a shows the charge–discharge curves at a current rate of C/20 during in situ XAS experiment. The V XANES spectrum during oxidation and reduction are normalized as described in previous literature.30 In the charge process below 4.3 V (#1 → #31) (Fig. 5b), a continuous edge shift to the higher energy can be observed, indicating a gradual oxidation from V3+ to Vx+ (4 < x < 5). During the charge process from 4.3 V to 4.8 V (#31 → #48) (Fig. 5c), V K-edge shifts further to the higher energy only a little with a significant increase of the pre-edge peaks, indicating a further oxidation of V ions and the increased distortion of V–O6 octahedra due to a stronger 3d–4p mixing and more overlap between metal 3d and ligand 2p orbitals.31 In the discharge process (Fig. 5d), the absorption edge shifting to lower energy and a decrease of pre-edge intensity demonstrate the reduction of V ions and recovery of the local structure (#48 → #80). Comparing the XANES spectra after one cycle with the pristine (Fig. S3a†), the edge position and the intensity of pre-edge can be fully recovered, indicating high reversibility of V oxidation and reduction during the first cycle, even for high cut-off voltage (4.8 V). This is different from most of layer-structured cathode materials, which cannot be fully recovered if they are overcharged.32 The magnitude plot of Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra for V K-edge (k3-weighted in k-space) are shown in Fig. S3b–d.† For the pristine LVP, three dominant peaks can be observed. The first peak at ∼1.6 Å is assigned to the single scattering path from V to the closest oxygen atoms (V–O bond in V–O6 octahedra), while the peak at ∼3.1 Å is attributed to the V–P distance between corner sharing P–O4 tetrahedra and V–O6 octahedra, and the peak at ∼4.1 Å is ascribed to V–V distance between two V–O6 octahedra. It should be noted that the FT-EXAFS spectra are not phase corrected so that the actual bond lengths were approximately 0.3–0.5 Å longer.33 Table S1† shows the least-square fitting results of the FT-EXAFS spectra for LVP at selected charge–discharge states. In the charge process, both V–O and V–P distance become shorter which is consistent with the decreased unit cell parameters as reported by Nazar et al.9 After charge to 4.8 V, the Debye–Waller factor increases significantly, indicating severe structure disorder, which could be the reason of high voltage hysteresis and increased charge-transfer resistance during high voltage charging (4.3 V → 4.8 V). In the discharge process, both V–O and V–P distances increase and can recover to the pristine values, indicating reversible local structure change. Surprisingly, both XANES and EXAFS data demonstrate a quite reversible V valence state and structure change of LVP during charge and discharge in the voltage window 3.0–4.8 V. It seems inconsistent with the capacity loss in this voltage window. Kang et al. also found the crystal structure of LVP can be fully recovered after one cycle in the voltage window 3.0–4.8 V by in situ X-ray diffraction.12 These solid evidences convince that LVP is a highly reversible framework during charge and discharge intrinsically, even at high cut-off voltage of 4.8 V.
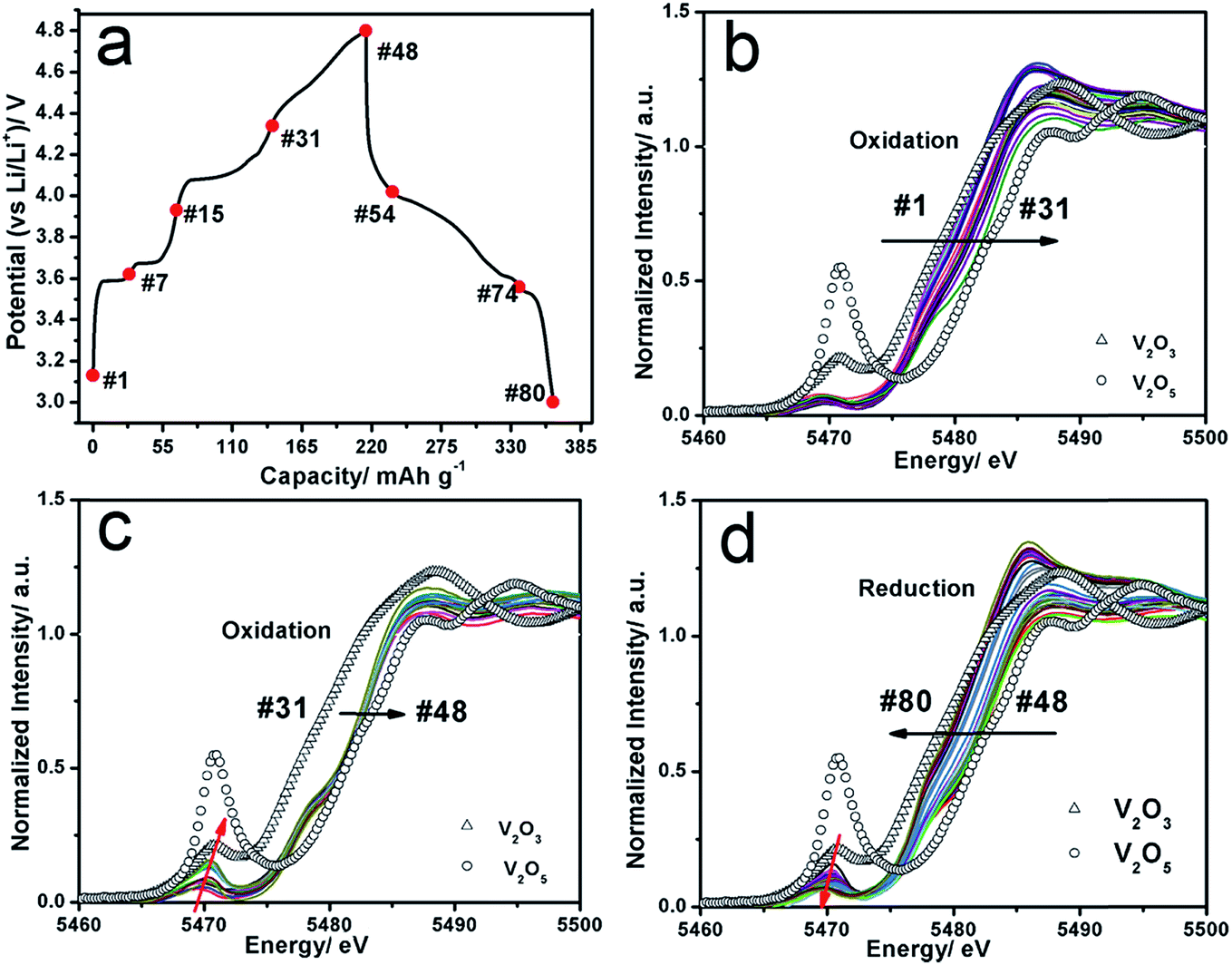 | ||
| Fig. 5 (a) Charge–discharge curves of LVP during in situ XAS experiment; (b and c) the corresponding V–K edge XANES spectra of LVP during the first charge process; (d) the corresponding V–K edge XANES spectra of LVP during the first discharge process. The standard spectra of V2O3 and V2O5 are plotted as reference. | ||
In order to find out the change of the cell components (cathode and anode) after multiple cycles in different voltage windows (3.0–4.3 V and 3.0–4.8 V), the coin cells were disassembled at the discharged state after 50 cycles. Their corresponding working electrodes were recovered for XRD characterization and the counter electrodes lithium foils were taken out for SEM and energy-dispersive X-ray spectroscopy (EDX) testing. Fig. S4† shows the XRD patterns of LVP electrode after 50 cycles. It is found that the peak intensity after cycling is comparable with the pristine LVP, indicates good crystallinity and structure reversibility. The lattice parameters of each pattern were obtained by FullProf _Suit match model, as shown in Table S2.† The lattice parameters of LVP electrode after 50 cycles are little smaller than those of the pristine LVP, indicating accumulated lattice contraction during multiple cycles. From V element mapping with EDX in SEM (Fig. 6), we found the trace of V element on the surface of lithium foil, indicating that some of V cations in LVP were dissolved into the electrolyte and shuttled to the lithium foil under electric fields. The V signal in the voltage window 3.0–4.8 V is higher than that in the voltage window 3.0–4.3 V. These results suggest that V dissolution into electrolyte is another factor for the capacity loss of LVP, especially during high voltage charge. Meanwhile, the structure stability of pristine Li3V2(PO4)3 at room temperature in air was also tested (Fig. S5†). Results reveal that Li3V2(PO4)3 is also sensitive to air (mainly H2O and O2) even at room temperature which can be slowly oxidized to Li3PO4 and VO(HPO4)·H2O. It implies that surface coating could be an effective approach to solve these problems.
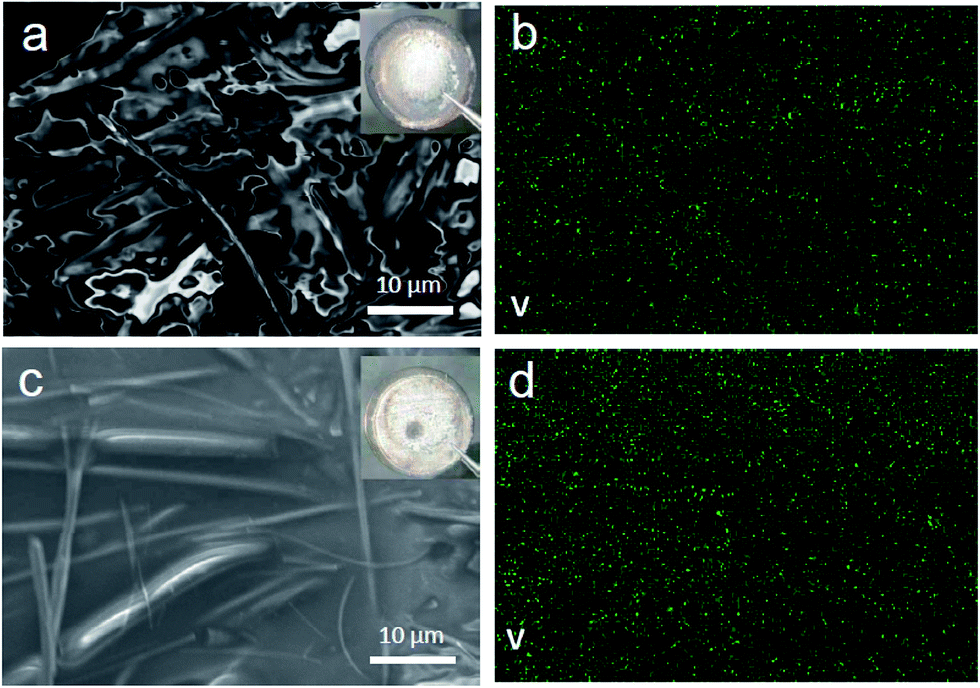 | ||
| Fig. 6 (a) SEM image and (b) V element mapping of the counter electrode lithium foil after LVP/Li half cell charge–discharged 50 cycles in the voltage window 3.0–4.3 V; (c) SEM image and (d) V element mapping of the counter electrode lithium foil after LVP/Li half cell charge–discharged 50 cycles in the voltage window 3.0–4.8 V. | ||
4. Conclusions
In summary, micro-sized LVP was synthesized via solid state reaction route. A “crystalline–amorphous–crystalline” phase transition pathway was revealed during the synthesis process from the precursors to the final LVP phase by using in situ XRD for the first time. The capacity retention behavior of LVP was evaluated in two different voltage windows: 3.0–4.3 V and 3.0–4.8 V. The apparent chemical diffusion coefficient of Li+ in LVP is obtained to be around 10−8 cm2 s−1, much higher than LiFePO4 (10−14 cm2 s−1). The capacity fading of LVP during charge and discharge in the voltage window 3.0–4.8 V is due to the parasitic reaction with electrolyte, V ion dissolution and poor kinetics for the third lithium extraction (LiV2(PO4)3 → V2(PO4)3), which lead to a higher voltage hysteresis and larger charge transfer resistance. Surprisingly, in situ X-ray absorption results show a quite reversible change of the valence state and local structure of V in the voltage window 3.0–4.8 V. It indicates that LVP is a promising multiple-electron-transfer cathode system with high structure reversibility intrinsically. Surface coating or modification, electrolyte optimization for high-voltage stability and doping for tuning its electronic structure could be effective strategies for upgrading the electrochemical performance of LVP.Acknowledgements
This work is supported by University of Electronic Science and Technology of China (Y02002010301080). We thank Dr Feng Wang and Dr Jianming Bai from Brookhaven National Laboratory for fruitful discussions.References
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS PubMed.
- K. Zaghib, A. Guerfi, P. Hovington, A. Vijh, M. Trudeau, A. Mauger, J. B. Goodenough and C. M. Julien, J. Power Sources, 2013, 232, 357–369 CrossRef CAS PubMed.
- H. Ohkawa, K. Yoshida, M. Saito, K. Uematsu, K. Toda and M. Sato, Chem. Lett., 1999, 1017–1018 CrossRef CAS.
- H. Huang, S. C. Yin, T. Kerr, N. Taylor and L. F. Nazar, Adv. Mater., 2002, 14, 1525–1528 CrossRef CAS.
- X. Rui, C. Li and C. Chen, Electrochim. Acta, 2009, 54, 3374–3380 CrossRef CAS PubMed.
- Z. Q. Liu, X. Y. Kang, C. F. Li, N. Hua, T. Wumair and Y. Han, J. Solid State Electrochem., 2012, 16, 1917–1923 CrossRef CAS.
- C. Jin, X. Zhang, W. He, Y. Wang, H. Li, Z. Wang and Z. Bi, RSC Adv., 2014, 4, 15332–15339 RSC.
- Z. P. Ma, G. J. Shao, X. Wang, J. J. Song and G. L. Wang, Ionics, 2013, 19, 1861–1866 CrossRef CAS.
- S. C. Yin, H. Grondey, P. Strobel, M. Anne and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 10402–10411 CrossRef CAS PubMed.
- S. Wang, Z. Zhang, S. Fang, L. Yang, C. Yang and S.-I. Hirano, Electrochim. Acta, 2013, 111, 685–690 CrossRef CAS PubMed.
- Q. L. Wei, Q. Y. An, D. D. Chen, L. Q. Mai, S. Y. Chen, Y. L. Zhao, K. M. Hercule, L. Xu, A. Minhas-Khan and Q. J. Zhang, Nano Lett., 2014, 14, 1042–1048 CrossRef CAS PubMed.
- J. Kang, V. Mathew, J. Gim, S. Kim, J. Song, W. B. Im, J. Han, J. Y. Lee and J. Kim, Sci. Rep., 2014, 4, 4047 Search PubMed.
- M. Choi, K. Kang, H.-S. Kim, Y. M. Lee and B.-S. Jin, RSC Adv., 2015, 5, 4872–4879 RSC.
- L. Fei, W. Lu, L. Sun, J. Wang, J. Wei, H. L. Chan and Y. Wang, RSC Adv., 2013, 3, 1297–1301 RSC.
- L. Q. Mai, S. Li, Y. F. Dong, Y. L. Zhao, Y. Z. Luo and H. M. Xu, Nanoscale, 2013, 5, 4864–4869 RSC.
- K. Nathiya, D. Bhuvaneswari, D. Nirmala and N. Kalaiselvi, RSC Adv., 2012, 2, 6885–6889 RSC.
- Z. Yang, J. Hu, Z. Chen, J. Zhong, N. Gu and N. Zhang, RSC Adv., 2015, 5, 17924–17930 RSC.
- M. Choi, K. Kang, H.-S. Kim, Y. M. Lee and B.-S. Jin, RSC Adv., 2015, 5, 4872–4879 RSC.
- M. Pivko, I. Arcon, M. Bele, R. Dominko and M. Gaberscek, J. Power Sources, 2012, 216, 145–151 CrossRef CAS PubMed.
- L. P. Wang, H. Li, M. Courty, X. J. Huang and E. Baudrin, J. Power Sources, 2013, 232, 165–172 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- Y.-N. Zhou, J.-J. Ding, K.-W. Nam, X. Yu, S.-M. Bak, E. Hu, J. Liu, J. Bai, H. Li and Z.-W. Fu, J. Mater. Chem. A, 2013, 1, 11130–11134 CAS.
- L. P. Wang, H. Li, X. J. Huang and E. Baudrin, Solid State Ionics, 2011, 193, 32–38 CrossRef CAS PubMed.
- Z. Chen, Y. Ren, Y. Qin, H. Wu, S. Ma, J. Ren, X. He, Y.-K. Sun and K. Amine, J. Mater. Chem., 2011, 21, 5604–5609 RSC.
- B. Zhang, X. Wang, H. Li and X. Huang, J. Power Sources, 2011, 196, 6992–6996 CrossRef CAS PubMed.
- S. C. Yin, P. S. Strobel, H. Grondey and L. F. Nazar, Chem. Mater., 2004, 16, 1456–1465 CrossRef CAS.
- K. Zhong, X. Xia, B. Zhang, H. Li, Z. Wang and L. Chen, J. Power Sources, 2010, 195, 3300–3308 CrossRef CAS PubMed.
- X. H. Rui, N. Ding, J. Liu, C. Li and C. H. Chen, Electrochim. Acta, 2010, 55, 2384–2390 CrossRef CAS PubMed.
- K. Tang, X. Q. Yu, J. P. Sun, H. Li and X. J. Huang, Electrochim. Acta, 2011, 56, 4869–4875 CrossRef CAS PubMed.
- Y. N. Zhou, J. Ma, E. Hu, X. Yu, L. Gu, K.-W. Nam, L. Chen, Z. Wang and X. Q. Yang, Nat. Commun., 2014, 5, 5381 CrossRef PubMed.
- A. Gutierrez, R. Qiao, L. Wang, W. Yang, F. Wang and A. Manthiram, Chem. Mater., 2014, 26, 3018–3026 CrossRef CAS.
- L. Liu, L. Chen, X. Huang, X.-Q. Yang, W.-S. Yoon, H. Lee and J. McBreen, J. Electrochem. Soc., 2004, 151, A1344–A1351 CrossRef CAS PubMed.
- Y. N. Zhou, M. Sina, N. Pereira, X. Yu, G. G. Amatucci, X. Q. Yang, F. Cosandey and K. W. Nam, Adv. Funct. Mater., 2015, 25, 696–703 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11510h |
| This journal is © The Royal Society of Chemistry 2015 |