DOI:
10.1039/C5RA11495K
(Paper)
RSC Adv., 2015,
5, 70271-70281
A novel triazine-aryl-bis-indole derivative inhibits both phosphodiesterase IV and expression of cell adhesion molecules†
Received
16th June 2015
, Accepted 5th August 2015
First published on 10th August 2015
Abstract
Asthma, like many inflammation related disorders, has a complex etiology. Drugs targeting multiple pathways may prove more efficacious in these complex disorders. Cyclic 3′,5′-adenosine monophosphate (cAMP) phosphodiesterase IV (PDE IV) is one of the validated targets in bronchial asthma and despite availability of some therapeutic molecules targeting PDE IV, molecules with better properties are desired. Eosinophil/neutrophil infiltration into lung may also be an important component of bronchial asthma in which increased expression of epithelial cell adhesion molecules may play an important role. This study describes the synthesis of a novel class of compounds ‘triazine-aryl-bis-indoles’ having a catechol derived structure constituting a part of ‘triazine’ and a part of ‘bis-indole’ moiety on it. This class of molecules potently inhibited both phosphodiesterase IV and expression of cell adhesion molecules ICAM-1 and VCAM-1. The best molecule of this class (compound 11) inhibited PDE IV activity in vitro, with an IC50 value of 14 μM compared to 12.7 μM for an existing drug rolipram. The compound 11 not only stabilized the cAMP level in human lung epithelial cells (L132) following stimulation with forskolin, but also inhibited TNF-α induced expression of cell adhesion molecules such as ICAM-1 and VCAM-1 in human umbilical vein epithelial cells (HUVECs). It also significantly inhibited the adhesion of human neutrophils to the endothelial monolayer (IC50 = 17.86 μM) in a dose dependent manner. Its absolute bioavailability (in mice) was found to be 70% and its toxicity and pharmacokinetic profiles are excellent. The dual activity of this class of molecules suggests that this class of molecules could have broad therapeutic applications in neutrophil dominant diseases such as severe asthma, COPD and acute lung injury.
Introduction
The design and development of novel phosphodiesterase (PDE) inhibitors in therapeutic applications have gained attention during the last decade.1–5 The PDEs are a superfamily of enzymes that catalyze the breakdown of cAMP and/or cyclic guanosine monophosphate (cGMP) to their inactive forms. The potential for selective PDE inhibitors to be used as therapeutic agents for various diseases was predicted earlier.6 PDE 4 is the main selective cAMP-metabolizing enzyme in inflammatory and immune cells. The role of cAMP as a second messenger is well established and it modulates the response of immune cells to a variety of stimuli. Elevation of cAMP has generally been associated with inhibition of lymphocyte activity.6 The elevation of cAMP levels leads to the suppression of the synthesis and release of pro-inflammatory signals, cytokines and inhibits the production of reactive oxygen species.7–9
Indoles, bis-indoles and octahydro indoles are very common structural skeletons of several biologically active compounds.10,11 The indole ring system is a very important component in many synthetic pharmaceuticals12,13 and it is worth to note that the World Drug Index contains 74 indole derivatives as drug molecules. Octahydro indole is a part structure of mesembrine, an alkaloid with PDE 4 inhibitory activity. Furthermore, the indole 3-acetic acid and its derivatives are commonly used as building blocks for the synthesis of pharmaceutically important molecules.14 Likewise, the 1,3,5-triazine skeleton is implicated in a variety of therapeutic activities and some triazine derivates have shown anti-asthmatic activity.15
In the present study, we described the synthesis of a novel ‘triazine-aryl-bis-indole’ derivative which inhibits phosphodiesterase activity in vitro and stabilized cAMP in vivo and demonstrated the anti-inflammatory activity of one of its potent derivative and thus these derivatives might be useful for the development of potential bronchodilator and/or anti-inflammatory agents for various airway diseases.
Results
Synthesis of triazine derivatives
The compounds described in this study were prepared following a general synthetic scheme using trichloro triazine as the starting material. Accordingly, trichloro triazine 1 on reacting with either cyclopropylamine or aniline under basic conditions (1 N NaOH/acetone/0–40 °C/2 h) furnished the corresponding disubstituted chloro 1,3,5-triazine 2 (92%) and 3 (98%) in excellent yields. Next, etherification (vanillin or isovanillin/K2CO3/DMF/70 °C/6 h) of compound 2 with vanillin and isovanillin independently led to triazine-aryl ethers 4 (74%) and 5 (82%) respectively, while compound 3 under similar conditions gave 6 (87%) and 7 (87%) (Scheme 1).
 |
| | Scheme 1 Synthesis of triazine aryl ethers. | |
Firstly, triazine-aryl ether 4 on reaction with indole or indole-3-acetic acid in the presence of an acid at 60 °C for 8 h gave triazine-aryl-bis-indoles 8 (76%) and 9 (52%), respectively. Similarly triazine-aryl ether 5 upon reaction with indole or indole-3-acetic acid under similar reaction conditions afforded triazine-aryl-bis-indoles 10 (67%) and 11 (61%), respectively (Scheme 2).
 |
| | Scheme 2 Synthesis of triazine-aryl-bis indoles. | |
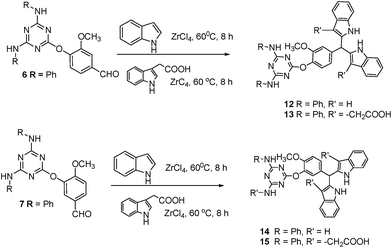
Next, the isomeric triazine-aryl ethers 6 and 7 on reaction with indole or indole-3-acetic acid resulted in the corresponding triazine-aryl-bis-indoles 12 (61%) and 13 (57%); 14 (66%) and 15 (64%) in comparable yields (Scheme 3). All the compounds were thoroughly characterized.
 |
| | Scheme 3 Synthesis of isomeric triazine-aryl-bis-indoles. | |
Triazine-aryl-bis-indole derivatives inhibit phosphodiesterase activity in vitro
PDE 4 enzyme preparations were purified from rat heart and incubated in the absence or presence of the compounds (10 μg ml−1) and breakdown of cAMP was quantified. As shown in Table 1, all the compounds inhibited PDE 4 activity which was comparable to the known PDE 4 inhibitor, rolipram. Out of 12 compounds tested 2 of them (compounds 11 and 15) showed moderately higher inhibition. We selected compound 11 for further investigation because of easy synthesis and higher yield. Dose response (0.1–20 μg ml−1) experiments showed that the IC50 value of compound 11 was higher than that of rolipram (13.03 ± 0.34 μM compound 11 compared to 12.7 ± 0.23 μM for rolipram) (Fig. 1).
Table 1 Effect of triazine-aryl-bis-indole derivatives on cAMP dependent phosphodiesterase activitya
| Compound no. |
Name |
Dose |
Inhibition (%) |
| The enzyme preparations were incubated in the absence or presence of compounds 2–14 and breakdown of cAMP was monitored by β counter. cAMP dependent PDE activity was measured as described in the methods. |
| 2 |
IICT-TA42 |
10 μg ml−1 |
44 |
| 3 |
IICT-TA57 |
10 μg ml−1 |
53 |
| 4 |
IICT-TA43 |
10 μg ml−1 |
29 |
| 5 |
IICT-TA58 |
10 μg ml−1 |
30 |
| 8 |
IICT-TA44 |
10 μg ml−1 |
16 |
| 9 |
IICT-TA45 |
10 μg ml−1 |
42 |
| 10 |
IICT-TA66 |
10 μg ml−1 |
34 |
| 11 |
IICT-TA67 |
10 μg ml−1 |
60 |
| 12 |
IICT-TA59 |
10 μg ml−1 |
34 |
| 13 |
IICT-TA60 |
10 μg ml−1 |
31 |
| 14 |
IICT-TA77 |
10 μg ml−1 |
45 |
| 15 |
IICT-TA78 |
10 μg ml−1 |
73 |
| Rolipram |
|
10 μg ml−1 |
69 |
 |
| | Fig. 1 Inhibition of phosphodiesterase activity by ‘triazine-aryl-bis-indole’ compound in vitro. Enzyme preparations were incubated in the absence or presence of different concentrations (1–20 μg ml−1) of compound 11 or rolipram and breakdown of cAMP was monitored by β counter. Enzyme activity was expressed as percentage of inhibition considering CPM value of enzyme without inhibitor as 100. Data represents mean ± SEM of 3 experiments (N = 3). *Indicates significantly different vs. control without compound. | |
Isozyme specificity of compound 11
To check the specificity and selectivity on the inhibitory activity of compound 11, screening of a panel of PDE isozymes was conducted using high throughput analysis. Out of 13 isozymes screened, compound 11 specifically inhibited PDE 4 family. It also inhibited the non selective PDE 5 and 6 isozymes. However, no inhibitory activity was recorded with PDE isozymes -1B, -2A, -3A, -3B, -7A, -8A1, -10A1 and -11A4 as shown in Table 2.
Table 2 PDE isozymes specificity of compound 11a
| Isozymes |
IC50 (μM) |
Tissue expression16,17 |
| IC50 of each PDE isozyme was determined in the presence of increasing concentrations of compound 11 (0.1 to 10 μg ml−1) using high throughput platform as described in the text. ND, non-detectable. |
| PDE 1B |
ND |
Brain, lymphocytes |
| PDE 2A |
ND |
Brain, heart, platelets, liver, thymocytes |
| PDE 3A |
ND |
Heart, blood vessels |
| PDE 3B |
ND |
Adipocytes, hepatocytes, lymphocytes |
| PDE 4A |
9.81 |
Lung, immune cells, brain, blood vessels |
| PDE 4B1 |
10 |
-do- |
| PDE 4D2 |
10 |
-do- |
| Non selective PDE 5 |
10 |
Smooth muscles, platelets |
| PDE 6 non selective |
5.34 |
Retinal photoreceptors |
| PDE 7A |
ND |
Brain, lymphocytes, kidney, heart |
| PDE 8A1 |
ND |
Thyroid |
| PDE 10A1 |
ND |
Testes, brain |
| PDE 11A4 |
ND |
Prostate, skeletal muscle, heart, testes |
Stabilization of cellular cAMP by compound 11
When human lung epithelial cells (L132) were stimulated with forskolin, the level of cAMP increased significantly over time in the presence of compound 11 compared to control treatment (Fig. 2). These data confirm our previous results that compound 11 inhibited PDE 4 leading to accumulation of cellular cAMP.
 |
| | Fig. 2 Effect of compound 11 on cellular cAMP level. Cellular level of cAMP was measured in L132 cells pre-treated with the compound (14.8 μM) for 5 min followed by addition of forskolin (40 μM). The reactions were terminated by the addition of ice-cold 95% ethanol at different time periods. The cells were harvested and centrifuged to remove cell debris. Measurement of cAMP was conducted using enzyme immunoassay kit (GE healthcare) as described by the manufacturer. The experiment was repeated 3 times with different batch of cells in duplicate wells for each time point. *Denotes significantly different at P < 0.01 vs. corresponding time points in the absence of the compound. | |
Compound 11 inhibits TNF-α induced expression ICAM-1 and VCAM-1 on HUVEC cells
Since it has been earlier demonstrated that cAMP is crucial in inducing various cell adhesion molecules via various mechanisms, we wanted to determine the effect of compound 11 on TNF-α induced expression of cell adhesion molecules, such as ICAM-1 and VCAM-1. For this, HUVECs were pretreated with various concentrations of the compound 11 for 2 hours followed by induction with TNF-α for 16 hours. The cell surface expression of cell adhesion molecules was measured by ELISA. As shown in Fig. 3, compound 11 inhibited the adhesion of human neutrophils to the endothelial monolayer in a dose-dependent way.
 |
| | Fig. 3 Compound 11 inhibits TNF-α induced expression ICAM-1 and VCAM-1 on HUVECs. HUVECs were seeded in 96-well plates at a cell density of 2 × 104 cells per well. Cells were pretreated with various concentrations of compound 11 for 2 hours followed by induction with TNF-α for 16 hours for ICAM-1 (A) and VCAM-1 (B). The cell surface expression of cell adhesion molecules was measured by ELISA. Experiment was repeated 3 times. Asterisk mark indicate significantly different compared to control experiment in the absence of compound 11. P < 0.05. | |
Compound 11 inhibits the adhesion of neutrophils on TNF-alpha induced HUVEC cells
The cell adhesion molecules such as ICAM-1 and VCAM-1 are critical in the adhesion of neutrophils to endothelial cells in most of the neutrophil dominant diseases. To evaluate the functional significance of inhibition of adhesion molecule by compound 11, we next checked adhesion of neutrophils on TNF induced HUVEC cells. As shown in Fig. 4, compound 11 dose dependently inhibited the adhesion of human neutrophils to the endothelial monolayer (IC50 = 17.8 μM) (Fig. 4). These data indicate that there is a strong correlation between inhibition of adhesion molecule expression and inhibition of neutrophil adhesion to the endothelial mono layer by the compound.
 |
| | Fig. 4 Compound 11 inhibits the adhesion of neutrophils on TNF-alpha induced HUVECs. HUVECs were seeded in 96-well plates at a cell density of 2 × 104 cells per well. Cells were pretreated with various concentrations of compound 11 for 2 hours followed by induction with TNF-α for 6 hours. Human blood neutrophils were added onto the endothelial cells monolayer and allowed to adhere for one hour at 37 °C. The number of cells that remained adhered to the monolayer was estimated by measuring the peroxidase activity. *Significantly (P < 0.05) different compared to control experiment in the absence of compound 11. | |
Pharmacokinetics of compound 11
Concentration vs. time curves for compound 11 after oral (Fig. 5A) and i.v. (Fig. 5B) administration in mice were established to determine the pharmacokinetics and bioavailability of the molecule. The results as summarized in Table 3 showed that after oral administration (20 mg kg−1) the half-life (t1/2) of compound 11 (IICTTA67) was 4.26 h; AUC = 502.75 ng h ml−1; clearance (CL) = 663 ml min−1, and volume of distribution (VD) = 474 l. After i.v. bolus dosing of 10 mg kg−1, compound 11 showed AUC = 357.71 ng h−1 ml−1; t1/2 = 0.93 h; CL = 466 ml min−1; VD = 37.5 l. Absolute bioavailability F (oral) of compound 11 (in mice) was found to be 70.30% (Table 3).
 |
| | Fig. 5 Pharmacokinetic profile of compound 11. Pharmacokinetics profiles of compound 11 either by oral (A) administration (20 mg kg−1) or through intravenous (B) route (10 mg kg−1 i.v.). Blood samples were drawn from retro-orbital plexus at indicated times in heparinised tubes and centrifuged (3500 rpm × 10 minutes) to obtain the plasma. Concentration–time curves for compound 11 were established and pharmacokinetic parameters were calculated as described in the experimental biology section. Values are mean ± SEM (n = 6). | |
Table 3 Pharmacokinetics for compound 11 in rata
| |
Oral |
i.v. |
| No Cmax and tmax in i.v. route. |
| Cmax (ng ml−1) |
107 |
— |
| tmax (hr) |
2.0 |
— |
| t1/2 (hr) |
4.26 |
0.93 |
| AUC (ng hr ml−1) |
502.75 |
357.71 |
| CL (ml min−1) |
663 |
466 |
| VD (l) |
474 |
37.5 |
Cytotoxicity of compound 11
The 3T3 Neutral Red Dye Uptake (NRU) cytotoxicity assay was conducted in the presence of increasing concentrations of compound 11 (Fig. 6). The derived IC50 of compound 11 was found to be 48 μM indicating that a reasonably high concentration of the compound is necessary for inducing cell death in vitro.
 |
| | Fig. 6 Determination of IC50 of compound 11 on 3T3 cells using the NRU cytotoxicity assay. Cytotoxicity is expressed as a concentration dependent reduction of the uptake of NR after compound 11 exposure, thus providing a sensitive, integrated signal of both cell integrity and growth inhibition. | |
Discussion
In this study, we demonstrate the synthesis of triazine-aryl-bis-indole derivatives and their effect on phosphodiesterase enzyme, an important target in the pathogenesis of asthma and related respiratory disorders. The indole and its various derivatives such as bis-indoles and octahydro indoles seem to be potent biologically active compounds.10,11 The indole moiety has been found not only in various natural molecules like lysergic acid, bufotenin, serotonin but also important for drug discovery research. These indole derivatives have been found to have anti-cancer, anti-oxidant, anti-inflammatory and antimicrobial activities.18 Importantly, indole derivatives have also shown to act as long acting β2adrenoceptor agonists similar to salmeterol.19 Since, bis-indoles have been found to exist in various nutritional components such as cruciferous vegetables18,20,21 it is likely that bis-indoles may be highly bioavailable and safer. The natural and unnatural analogs of bis-indole derivatives have potent anticancer activities.18 In addition to these, triazine derivatives also have anti-eosinophilic and anti-asthma activity.22 Triazine aryl-bis-indole derivatives are synthesized by the dialkylation of triazine followed by etherification with vanillin–isovanillin. Furthermore, these triazine aryl ethers were derivatised by the condensation of indole of indole acetic acid in the presence of TCT under mild conditions for achieving the target compounds. These compounds were evaluated for inhibitory activity against various isoforms of PDE enzyme. These evidences indicate that triazine aryl-bis-indole derivatives could be potent in various diseases.
The cyclic nucleotide PDEs hydrolyze cAMP and cGMP to convert into their inactive analogues. The potential for selective PDE inhibitors as therapeutic agents was predicted earlier. Thus, it has been focused to discover novel PDE 4 inhibitors for using in various diseases. In this context, indole derivatives also have been shown to have PDE 4 inhibitory activity.23 In this study, we synthesize effective indole derivatives and checked their PDE inhibitory activities. Among various PDE isoenzymes known so far, PDE 4 is crucial as it is predominantly present in various inflammatory cells.23 It is important to note that only PDE 4, 7 and 8 are associated with the metabolism of cAMP. The role of cAMP has been implicated in bronchial asthma pathogenesis.24 Because PDE inhibitors prevent breakdown of cAMP they act as bronchodilators by maintaining high level of this second messengers in bronchial cells.25 Abundance of PDE 4 in lung tissue has been reported earlier.26 PDE 4 has four distinct genes, PDE 4A, PDE 4B, PDE 4C and PDE 4D27 with specificity to cAMP and thus have become potential therapeutic targets. The importance of PDE 4 as a therapeutic target for respiratory disorders has been supported further by the recent approval of roflumilast, an inhibitor of PDE 4, by FDA, for the treatment of chronic obstructive pulmonary disease.28–31 In this report, we describe 12 compounds having appreciable PDE 4 activity in vitro. Out of which 2 had better activity. In vitro inhibitory activity of this compound on PDE 4 is further supported by our data on stabilization of intracellular cAMP in lung epithelial cells. For synthesis and better yield we had detailed investigation with isozyme specificity and other relevant studies with one compound (compound 11). This compound strongly inhibits PDE 4A, PDE 4B and PDE 4D isozymes which are mainly expressed in lung and immune cells.16,17 Compound 11 also inhibits non-selective PDE 5 and PDE 6 isozymes. However, these isozymes are not expressed in lung or immune cells.16,17 Thus it appears that non selective inhibitory activities of compound 11 may not have any influence in asthma pathogenesis. Interestingly, this compound does not inhibit isozymes prevalent in brain (PDE 1A, PDE 1B and PDE 7) or heart (PDE 2A, PDE 3A, PDE 7A and PDE 11A4) which strengthens its potential selective application against respiratory diseases.
The second messenger cAMP controls many cellular functions and it is well established that an elevated cAMP level can inhibit various inflammatory processes. Thus, inhibitors of enzymes that catalyze cAMP hydrolysis would seem to be good candidates to treat inflammatory conditions.9 It is known that cAMP reduces inflammatory signaling by inhibiting TNF-α induced cell adhesion molecules through inhibiting either NF-KB or reactive oxygen species.32,33 On the other hand, cell adhesion molecules are crucial in various inflammatory diseases. Recruitment of inflammatory cells including eosinophils in the lung tissue plays an important role in asthma pathogenesis.34 The recruitment of various immune cells into the site of the injury depends on the timely expression of cell adhesion molecules on vascular endothelial cell surface and adhesion of immune cells such as neutrophils. Migration of these inflammatory cells is largely regulated by the expression of adhesion molecules such as ICAM-1 and VCAM-1 either on the epithelial or on the endothelial cell surface.35–37 Because of anti-inflammatory activities, PDE inhibitors are known to inhibit cellular trafficking, production of oxygen species and cell adhesion molecule expression.8 Consistently, we also show significant inhibition of adhesion molecules in HUVEC cells by our compound suggesting its multipronged efficacy in inhibiting asthma pathogenesis. These data indicate the potential of this compound as a therapeutic molecule against bronchial asthma.
To determine whether the compound 11 is orally active, evaluation of the in vivo pharmacokinetic parameters was performed. With oral dosing maximum plasma concentration of compound 11 attains rapidly within 2 h. It also has a higher t1/2 value with moderately higher clearance and distribution volume with oral dosing compared to i.v. administration. Most of the compound was eliminated by 24 h with elimination half life of about 4.25 h. Oral bioavailability was found to be 70%. These data suggest that compound 11 is orally bioavailable with good pharmacokinetic profile.
The cytotoxic IC50 of compound 11 is approximately 5–10 fold greater than other anti-inflammatory compounds such as ursolic acid38,39 and TiO2.40 However, the cytotoxic IC50 of several PDE 4 inhibitors designed elsewhere41 were >80 μM after a 24 h assay. Notably, the duration of compound 11 exposure was 48 h and this prolonged exposure is likely to be associated with a pronounced occurrence of a cumulative cytotoxic effect. Despite cumulative cytotoxic effect, the IC50 of compound 11 offers ∼2 fold margin in comparison to our observed IC50 of compound 11 inhibiting ICAM (Fig. 3a), VCAM (Fig. 3b), neutrophil adhesion molecule (Fig. 4), as well as >3 fold safe margin over PDE 4 inhibition (14 μM). Hence, the compound 11 is toxicologically safe enough to merit clinical evaluation and use.
Thus, triazine-aryl-bis-indole derivatives are novel structure with ability to inhibit hydrolysis of cAMP in in vivo conditions in association with inhibition of adhesion molecules (Fig. 7). This compound may potentiate bronchodilation due to sustenance of intracellular cAMP. Furthermore, it may act as an anti-inflammatory by inhibiting adhesion molecules (Fig. 7).
 |
| | Fig. 7 Schematic representation of probable mechanism of action of compound 11. Scheme depicts conversion of ATP to cAMP by adenylate cyclase (AC) which is normally hydrolyzed to adenosine monophosphate (AMP) by endogenous phosphodiesterase (PDE). Compound 11 inhibits (┬) PDE and helps maintaining increased (↑) level of cAMP which is known to cause bronchodilation.23,24,27–30 Compound 11 also inhibits (┬) cell adhesion molecules, ICAM and VACM which likely (broken arrow) to help bronchodilation by preventing (down dashed arrow) ongoing inflammatory process in respiratory diseases. | |
Experimental procedure
Synthesis of triazine-aryl-bis-indoles
Column chromatography was performed on silica gel, Acme grade 60–120 mesh. Unless stated otherwise, all reagents were purchased from commercial sources (Sigma-Aldrich Chemicals, St. Louis, MO, USA) and used without additional purification. 1H NMR and 13C NMR spectra were recorded either on a Bruker 300 or Varian VXR 400 or Varian VXR 500 in CDCl3 or CDCl3 + DMSO mixture as solvent with TMS as reference unless otherwise indicated. Unless stated otherwise, HRMS spectra were recorded on a QTOF analyser (QSTAR XL, Applied Biosystems/MDS Sciex) at NCMS-IICT, Hyderabad. Unless stated otherwise, elemental Analysis was carried on a Vario Micro Cube Elementar at Analytical Chemistry Division IICT, Hyderabad. The software ACD/Name Version 1.0, developed by M/s Advanced Chemistry Development Inc., Toronto, Canada, assisted nomenclature used in the Experimental section. Unless stated otherwise, all the reactions were performed under inert atmosphere. Representative NMR and LCMS spectra of the compounds are presented in ESI† file.
6-Chloro-N2,N4-dicyclopropyl-1,3,5-triazine-2,4-diamine 2. A fine slurry prepared from cyanuric chloride 1 (5.0 g, 27.17 mmol) by the addition of acetone (25 ml) was stirred at 40 °C for 30 min. The reaction mixture was cooled to 0 °C and treated with cyclopropylamine (3.09 g, 54.34 mmol). It was stirred at the same temperature for 1 h and neutralized with aq. 1 N NaOH till the reaction mixture becomes basic. It was then heated to 40 °C and stirred at this temperature for 2 h. The reaction mixture was filtered, washed with acetone (3 × 25 ml), evaporated solvent and dried under vacuum to furnish 2 (5.6 g, 92%) as a white solid. Mp 197–199 °C; IR (KBr): 3239, 3094, 3013, 2935, 1609, 1542, 1350, 1279, 1018, 807, 740 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.08–6.62 (m, 2H), 3.26–2.63 (m, 2H), 0.85–0.49 (m, 8H); 13C NMR (75 MHz, CDCl3): δ 167.9, 166.7, 166.5, 23.4, 23.3, 6.0, 5.8; HRMS (ESI): m/z calculated for C9H13ClN5: 226.0854 (M + 1)+; found: 226.0852.
6-Chloro-N2,N4-diphenyl-1,3,5-triazine-2,4-diamine 3. A fine slurry of cyanuric chloride 1 (5.0 g, 27.17 mmol) and acetone (25 ml) was stirred at 40 °C for 30 min. The reaction mixture was cooled to 0 °C and treated with aniline (5.05 g, 54.34 mmol) and worked up as described for 2 to furnish 3 (7.45 g, 98%) as a white solid. Mp 210–212 °C; IR (KBr): 3291, 3053, 2924, 2850, 1687, 1609, 1512, 1442, 1276, 1124, 746 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 9.30 (br. s, 1H), 7.56 (d, 2H, J = 7.5 Hz), 7.39 (s, 1H), 7.24–7.18 (m, 5H), 6.98 (t, 2H, J = 7.3 Hz), 163.4, 137.5, 127.8, 122.9, 120.4; HRMS (ESI): m/z calculated for C15H13N5Cl [M + H]+: 298.0854; found: 298.0852.
4-[4,6-Bis-cyclopropylamino-1,3,5-triazin-2-yl-oxy]-3-methoxy benzaldehyde 4. A mixture of 2 (5.0 g, 22.1 mmol), vanillin (4.04 g, 26.6 mmol) and K2CO3 (9.17 g, 66.48 mmol) in DMF (30 ml) was heated at 70 °C for 6 h. The reaction mixture was cooled to room temperature, diluted with water (75 ml) and extracted with EtOAc (4 × 50 ml). The combined organic layers were washed with water (50 ml), brine (50 ml) and dried (Na2SO4). Evaporation of the solvent under reduced pressure and purification of the residue by column chromatography (silica gel, EtOAc in hexane 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) furnished 4 (5.58 g, 74%) as a white solid. Mp 138–140 °C; IR (KBr): 3313, 3093, 3008, 2850, 1690, 1604, 1542, 1352, 1282, 809, 780 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 9.95 (s, 1H), 7.78 (s, 1H), 7.49 (s, 1H), 7.31 (d, 1H, J = 6.6 Hz), 6.98–6.55 (m, 2H), 3.87 (s, 3H), 3.07–2.62 (m, 2H), 0.75–0.39 (m, 8H); 13C NMR (75 MHz, DMSO-d6): δ 191.9, 168.3, 167.7, 167.2, 151.9, 146.1, 134.9, 123.6, 111.8, 55.8, 23.3, 23.2, 5.9, 5.7; HRMS (ESI): m/z calculated for C17H20O3N5 (M + 1)+ 342.1560; found: 342.1577.
:7) furnished 4 (5.58 g, 74%) as a white solid. Mp 138–140 °C; IR (KBr): 3313, 3093, 3008, 2850, 1690, 1604, 1542, 1352, 1282, 809, 780 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 9.95 (s, 1H), 7.78 (s, 1H), 7.49 (s, 1H), 7.31 (d, 1H, J = 6.6 Hz), 6.98–6.55 (m, 2H), 3.87 (s, 3H), 3.07–2.62 (m, 2H), 0.75–0.39 (m, 8H); 13C NMR (75 MHz, DMSO-d6): δ 191.9, 168.3, 167.7, 167.2, 151.9, 146.1, 134.9, 123.6, 111.8, 55.8, 23.3, 23.2, 5.9, 5.7; HRMS (ESI): m/z calculated for C17H20O3N5 (M + 1)+ 342.1560; found: 342.1577.
4-[4,6-Bis-cyclopropylamino-1,3,5-triazin-2-yl-oxy]-4-methoxy benzaldehyde 5. A mixture of 2 (5.0 g, 22.1 mmol), isovanillin (4.04 g, 26.6 mmol) and K2CO3 (9.17 g, 66.51 mmol) in DMF (30 ml) using the procedure as described for 4 furnished 5 (6.2 g, 82%) as a white solid. Mp 189–191 °C; IR (KBr): 3297, 3255, 3008, 2847, 1605, 1575, 1506, 1363, 1280, 812, 729 cm−1; 1H NMR (500 MHz, CDCl3 + DMSO-d6): δ 9.88 (s, 1H), 7.75 (d, 1H, J = 8.3 Hz), 7.71–7.61 (m, 1H), 7.08 (d, 1H, J = 8.4 Hz), 5.97–5.58 (m, 2H), 3.89 (s, 3H), 2.94–2.48 (m, 2H), 0.81–0.39 (m, 8H); 13C NMR (75 MHz, CDCl3 + DMSO-d6): δ 189.6, 169.6, 167.9, 167.3, 156.5, 141.3, 129.6, 128.6, 123.0, 111.3, 55.5, 22.7, 6.3; HRMS (ESI): m/z calculated for C17H20N5O3: 342.1566 (M + 1)+; found 342.1574.
4-[4,6-Bis-anilino-1,3,5-triazin-2-yl-oxy]-3-methoxy benzaldehyde 6. Using a mixture of 3 (5 g, 16.79 mmol), vanillin (3.06 g, 20.15 mmol) and K2CO3 (9.17 g, 66.51 mmol) in DMF (30 ml) and adopting the procedure as described for 4 gave 6 (6.05 g, 87%) as a white solid. Mp 178–180 °C; IR (KBr): 3292, 3110, 1684, 1623, 1581, 1525, 1443, 1400, 1355, 1287, 1156, 1112, 751; 1H NMR (75 MHz, DMSO-d6): δ 10.00 (s, 1H), 8.98 (br. s, 2H), 7.99 (s, 1H), 7.81–7.43 (m, 5H), 7.55–6.9 (m, 7H), 3.85 (s, 3H); 13C NMR (75 MHz, DMSO-d6): δ 190.3, 164.5, 161.6, 151.5, 145.7, 137.9, 133.7, 127.4, 123.5, 122.0, 119.7, 110.1, 55.0; HRMS (ESI): m/z calculated for C23H20O3N5 [M + H]+: 414.1560; found: 414.1526.
4-[4,6-Bis-anilino-1,3,5-triazin-2-yl-oxy]-4-methoxy benzaldehyde 7. A mixture of 3 (5 g, 16.79 mmol), isovanillin (3.06 g, 20.15 mmol) and K2CO3 (6.95 g, 50.38 mmol) in DMF (30 ml) according to the procedure as described for 4 gave 7 (6.01 g, 87%) as a white solid. Mp 148–150 °C; IR (KBr): 3405, 2925, 1698, 1612, 1574, 1505, 1440, 1391, 1348, 1210, 1125, 746 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 9.89 (br. s, 1H), 8.75 (s, 2H), 7.70 (d, 1H, J = 2.0 Hz), 7.59–7.47 (m, 4H), 7.29–6.99 (m, 8H), 3.87 (s, 3H); 13C NMR (75 MHz, DMSO-d6): δ 190.3, 164.5, 161.6, 151.5, 145.7, 137.9, 137.7, 127.4, 123.5, 122.7, 122.0, 119.7, 110.1, 55.0; HRMS (ESI): m/z calculated for C23H20O3N5 [M + H]+: 414.1560; found: 414.1558.
2,2′-[2,2′-4-(4,6-Bis-cyclopropylamino)-1,3,5-triazin-2-yl-oxy-3-methoxyphenyl]-methylene-bis-1H-indole 8. To a stirred solution of 4 (1.0 g, 2.9 mmol) and indole (0.69 g, 5.8 mmol) in CH3CN (5 ml), catalytic amount of cyanuric chloride was added and heated at 60 °C for 8 h. The reaction mixture was cooled to room temperature, diluted with water (25 ml) and extracted with EtOAc (4 × 50 ml). The combined organic layers were washed with water (50 ml), brine (50 ml) and dried (Na2SO4). Evaporation of the solvent under reduced pressure and purification of the residue by column chromatography (silica gel, EtOAc in hexane 1:4) afforded 8 (1.25 g, 76%) as a pale brown solid. Mp 185–187 °C; IR (KBr): 3414, 3267, 3008, 1586, 1505, 1356, 1207, 1147, 810, 749 cm−1; 1H NMR (300 MHz, CDCl3 + DMSO-d6): δ 10.28 (s, 2H), 7.73 (m, 2H), 7.33 (d, 4H, J = 7.9 Hz), 7.12–7.00 (m, 3H), 7.00–6.74 (m, 5H), 6.63 (s, 1H), 5.8 (s, 1H), 3.66 (s, 3H), 2.90–2.53 (m, 2H), 0.74–0.34 (m, 8H); 13C NMR (75 MHz, CDCl3 + DMSO-d6): δ 169.7, 167.9, 167.36, 150.7, 142.2, 139.1, 136.3, 126.2, 123.2, 121.8, 120.6, 118.8, 117.8, 112.6, 110.9, 55.25, 30.8, 22.9, 6.0; HRMS (ESI): m/z calculated for C33H32N7O2: 558.2612 (M + 1)+; found: 558.2587.
2,2′-[2,2′-4-(4,6-Bis-cyclopropylamino)-1,3,5-triazin-2-yl-oxy-3-methoxyphenyl]-methylene-bis-(1H-indole-3,3-diyl)-diacetic acid 9. A mixture of 4 (1.0 g, 2.9 mmol) and indole 3-acetic acid (1.02 g, 5.86 mmol) in CH3CN (5 ml) was treated with a catalytic amount of ZrCl4 and heated at 60 °C for 8 h. The reaction mixture was cooled to room temperature, diluted with water (25 ml) and extracted with EtOAc (4 × 50 ml). The combined organic layers were washed with water (50 ml), brine (50 ml) and dried (Na2SO4). Evaporation of the solvent under reduced pressure and purification of the residue by column chromatography (silica gel, EtOAc in hexane 1:4) afforded 9 (1.02 g, 52%) as a pale brown solid. Mp 254–256 °C; IR (KBr): 3429, 3357, 3007, 2924, 1708, 1584, 1509, 1408, 1352, 1204, 809, 743 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 10.5 (br. s, 2H), 7.8 (d, 3H, J = 6.4 Hz), 7.5 (d, 2H, J = 7.5 Hz), 7.2 (d, 2H, J = 7.7 Hz), 7.12–6.9 (m, 7H), 6.8 (s, 1H), 6.1 (s, 1H), 3.63 (s, 3H), 3.62 (s, 2H), 3.3 (s, 2H), 2.92–2.5 (m, 2H), 0.62 (m, 8H); 13C NMR (75 MHz, DMSO-d6): δ 173.1, 169.33, 167.2, 151.0, 139.6, 138.6, 135.5, 135.2, 127.7, 122.6, 120.9, 118.6, 118.1, 111.0, 105.5, 55.6, 30.0, 23.4, 5.9; HRMS (ESI): m/z calculated for C37H36N7O6: 674.2721 (M + 1)+; found: 674.2716.
2,2′-[2,2′-3-(4,6-Bis-cyclopropylamino)-1,3,5-triazin-2-yl-oxy-4-methoxyphenyl]-methylene-bis-1H-indole 10. To a stirred solution of 5 (1.0 g, 2.9 mmol) and indole (0.68 g, 5.85 mmol) in CH3CN (5 ml) adopting the procedure as described for 8 gave 10 (1.1 g, 67%) as a pale brown solid. Mp 176–178 °C; IR (KBr): 3410, 3007, 2923, 2852, 1586, 1506, 1354, 1270, 1129, 808, 747 cm−1; 1H NMR (500 MHz, DMSO-d6): δ 10.33 (s, 2H), 7.29–7.19 (m, 4H), 7.08 (d, 1H, J = 7.1 Hz), 7.01–6.92 (m, 3H), 6.84–6.66 (m, 7H), 5.67 (s, 1H), 3.6 (s, 3H), 2.83–2.45 (m, 2H), 0.45 (m, 8H); 13C NMR (75 MHz, DMSO-d6): δ 167.77, 168.31, 149.3, 136.4, 126.4, 123.3, 120.7, 118.9, 118.08, 117.97, 111.29, 55.56, 38.5, 22.9, 5.9; HRMS (ESI): m/z calculated for C33H32N7O2: 558.2612 (M + 1)+; found: 558.2642.
2,2′-[2,2′-3-(4,6-Bis-cyclopropylamino)-1,3,5-triazin-2-yl-oxy-4-methoxyphenyl]-methylene-bis-(1H-indole-3,3-diyl)-diacetic acid 11. A mixture of 5 (1.0 g, 2.9 mmol) and indole 3-acetic acid (1.02 g, 5.85 mmol) in CH3CN (5 ml) using the procedure as described for 9 furnished 11 (1.2 g, 61%) as a pale brown solid. Mp 204–206 °C; IR (KBr): 3387, 2921, 2851, 1707, 1591, 1511, 1356, 1272, 1023, 810, 744 cm−1; 1H NMR (500 MHz, DMSO-d6): δ 10.4 (s, 2H), 7.77 (d, 2H, J = 6.2 Hz), 7.48 (d, 2H, J = 7.5 Hz), 7.26 (d, 2H, J = 7.3 Hz), 7.13–6.91 (m, 6H), 6.8 (d, 1H, J = 8.8 Hz), 6.5 (br. s, 2H), 6.11 (s, 1H), 3.74 (s, 3H), 3.61 (s, 2H), 2.82–2.5 (m, 2H), 0.77–0.39 (m, 8H); 13C NMR (75 MHz, DMSO-d6): δ 173.2, 169.68, 168.33, 167.36, 150.2, 140.9, 135.4, 127.7, 125.6, 122.5, 120.9, 118.2, 118.5, 110.9, 105.5, 55.7, 38.72, 29.6, 23.4, 5.9; HRMS (ESI): m/z calculated for C37H36N7O6: 674.2721 (M + 1)+; found: 674.2716.
2,2′-[2,2′-4-(4,6-Bis-anilino)-1,3,5-triazin-2-yl-oxy-3-methoxyphenyl]-methylene-bis-1H-indole 12. Using a stirred solution of 6 (1.0 g, 2.42 mmol) and indole (0.56 g, 4.84 mmol) in CH3CN (5 ml) adopting the procedure as described for 8 furnished 12 (1.0 g, 61%) as a pale brown solid. Mp 180–182 °C; IR (KBr): 3396, 3052, 2927, 1609, 1576, 1503, 1441, 1391, 1346, 1200, 1121, 1031, 744; 1H NMR (75 MHz, DMSO-d6): 10.78 (m, 2H), 9.30 (br. s, 2H), 7.92–6.63 (m, 23H), 5.86 (s, 1H), 3.37 (br. s, 3H); 13C NMR (75 MHz, DMSO-d6): δ 170.3, 165.1, 150.6, 143.5, 139.0, 136.4, 128.1, 126.5, 123.3, 122.3, 120.8, 120.2, 118.9, 118.1, 111.3, 99.6, 59.6, 55.5; HRMS (ESI): m/z calculated for C39H32O2N7 [M + H]+: 630.2612; found: 630.2563.
2,2′-[2,2′-4-(4,6-Bis-anilino)-1,3,5-triazin-2-yl-oxy-3-methoxyphenyl]-methylene-bis-(1H-indole-3,3-diyl)-diacetic acid 13. A mixture of 6 (1.0 g, 2.41 mmol) and indole 3-acetic acid (0.84 g, 4.83 mmol) in CH3CN (5 ml) using the procedure as described for 9 gave 13 (1.02 g, 57%) as a pale brown solid. Mp 240–242 °C; IR (KBr): 3364, 2924, 1707, 1611, 1576, 1514, 1443, 1413, 1352, 1299, 1204, 1123, 1027, 747; 1H NMR (500 MHz, DMSO-d6): δ 9.78–9.53 (m, 2H), 8.46 (br. s, 2H), 7.61–7.28 (m, 10H), 7.16–6.86 (m, 13H), 6.73 (s, 1H), 3.60 (br. s, 7H); 13C NMR (75 MHz, DMSO-d6): δ 173.1, 171.8, 170.2, 165.1, 151.0, 139.6, 139.1, 139.0, 135.6, 135.2, 128.1, 127.6, 122.4, 120.9, 120.3, 118.5, 118.1, 113.4, 111.1, 105.4, 59.6, 55.6, 29.9; HRMS (ESI): m/z calculated for C43H36O6N7 [M + H]+: 746.2721; found: 746.2721.
2,2′-[2,2′-3-(4,6-Bis-anilino)-1,3,5-triazin-2-yl-oxy-4-methoxyphenyl]-methylene-bis-1H-indole 14. To a stirred solution of 7 (1.0 g, 2.4 mmol) and indole (0.56 g, 4.8 mmol) in CH3CN (5 ml) adopting the procedure as described for 8 furnished 14 (1.0 g, 66%) as a pale brown solid. Mp 165–168 °C; IR (KBr): 3379, 2924, 1614, 1579, 1513, 1443, 1399, 1268, 1129, 743; 1H NMR (300 MHz, DMSO-d6): δ 9.84 (br. s, 2H), 8.74 (br. s, 2H), 7.82–7.46 (m, 5H), 7.44–7.32 (m, 4H), 7.31–6.63 (m, 14H), 5.89 (s, 1H), 3.71 (s, 3H); 13C NMR (75 MHz, DMSO-d6): δ 170.0, 164.7, 148.9, 140.3, 138.2, 136.9, 136.0, 127.5, 126.0, 125.2, 123.0, 122.1, 120.3, 119.8, 118.6, 117.5, 115.0, 111.4, 110.5, 59.3, 55.0; HRMS (ESI): m/z calculated for C39H32O2N7 [M + H]+: 630.2612; found: 630.2604.
2,2′-[2,2′-3-(4,6-Bis-anilino)-1,3,5-triazin-2-yl-oxy-4-methoxyphenyl]-methylene-bis-(1H-indole-3,3-diyl)-diacetic acid 15. A mixture of 7 (1.0 g, 2.4 mmol) and indole 3-acetic acid (0.85 g, 4.8 mmol) in CH3CN (5 ml) using the procedure as described for 9 gave 15 (1.15 g, 64%) as a pale brown solid. Mp 188–190 °C; IR (KBr): 3262, 3092, 1607, 1575, 1521, 1445, 1388, 1226, 990, 805, 754, 703, 599; 1H NMR (300 MHz, CDCl3 + DMSO-d6): δ 9.93 (br. s, 2H), 8.27 (br. s, 2H), 7.69–7.34 (m, 8H), 7.24–6.99 (m, 15H), 6.06 (s, 1H), 3.63 (s, 3H), 3.25 (s, 4H); 13C NMR (75 MHz, CDCl3 + DMSO-d6): δ 188.7, 172.0, 168.8, 163.9, 155.4, 140.3, 137.7, 134.8. 134.1, 128.3, 126.7, 125.7, 122.1, 121.7, 121.1, 119.6, 119.0, 111.0, 109.9, 106.2, 104.0, 54.7, 54.3, 29.7; HRMS (ESI): m/z calculated for C43H36O6N7 [M + H]+: 746.2721; found: 746.2711.
Animal
Healthy Swiss mice of both the sexes (n = 25–30) were used for pharmacokinetic study. The animals were kept under controlled conditions (temp 26 ± 2 °C; relative humidity 50 ± 5%; 12 h light/dark cycle) and maintained on pelleted rodent diet (Ashirwad Industries Ltd Chandigarh, India). Water was provided ad libitum. Institutional Animal Ethics Committee approved the animal experiments. Animals were fasted for 16 hours before use.
Phosphodiesterase (PDE 4) assay
PDE enzyme preparations were performed from rat heart.42 Heart was excised from anesthetized rat and transfer into normal saline solution. Heart weighing 687 mg was taken and homogenized in 2.5 volumes of homogenization buffer containing 20 mM Tris–Cl, 50 mM NaCl, 2 mM EDTA, 0.1 mM PMSF, 1 mH DTT, protease inhibitors 1 μg μl−1. The homogenate was centrifuged at 500 × g for 5 minute at 4 °C. Then supernatant was further centrifuged at 40000 × g for 30 minute (ultra centrifugation). After centrifugation, supernatant was collected for phosphodiesterase assay. Protein concentration was estimated and for each assay, 2.5 μg protein (PDE enzyme preparation) was used. All compounds tested for PDE 4 activity were dissolved in DMSO.
Phosphodiesterase 4 assay was conducted with [3H] cAMP SPA enzyme assay (GE Amersham, UK) using 2.5 μg enzyme preparation for each well. Into each well 60 μl water, 10 μl assay buffer supplied with the kit, 10 μl compound (1–13), 10 μl PDE enzyme or homogenizing buffer (blank) and 10 μl radiolabel cAMP ([3H]cAMP, GE Amersham, UK) were added. To check the activity of the enzyme without inhibitor 10 μl DMSO was added in place of inhibitor to a separate well. The reaction mixtures were incubated for 30 min at 30 °C. The reaction was stopped by adding 50 μl SPA bead supplied with the kit. It was then mixed well and allowed to stand at room temperature for 20 min. Breakdown of cAMP in each tube was monitored by β counter. The reading in each tube was obtained in counts per minute (CPM). The reading of the blank tube (without enzyme) was subtracted from each tube to normalize the counts. The enzyme activity was calculated by considering the CPM reading without any inhibitor (compound) as 100.
High throughput phosphodiesterase profiling
To determine the inhibitory activity and selectivity of triazine-aryl-bis-indole derivative on the PDE superfamily high throughput profiling platform containing a panel of 13 isozymes was utilized (CEREP, France). Inhibitory activity of compound 11 was examined in 5 different concentrations (0.1, 1, 2, 5 and 10 μg ml−1). Inhibitory activity was expressed in terms of IC50 value obtained from the plot of 5 different concentrations in duplicates.
Measurement of cellular level of cAMP
Human lung epithelial cell line (L132) cells43,44 were grown to 70% confluence. After overnight serum starvation, the cells were pre-treated with the compound (10 μg ml−1) for 5 min followed by addition of forskolin, (40 μM, Sigma Aldrich) and the reactions were terminated by the addition of ice-cold 95% ethanol at different time periods. The cells were harvested and centrifuged to remove cell debris. The experiment was repeated on at least three separate occasions in duplicate wells for each time point. Measurement of cAMP was conducted using enzyme immunoassay kit (GE healthcare) as described by the manufacturer.
Measurement of ICAM-1 and VCAM-1
Cell-ELISA was used for measuring the expression of ICAM-1 and VCAM-1 on surface of endothelial cells (HUVEC) as described earlier.45 Briefly, HUVEC cells were incubated with or without compound 11 at desired concentrations for the required period, followed by treatment with TNF-α (10 ng ml−1) for 16 h. The cells were then fixed with 1.0% glutaraldehyde. Non-specific binding of antibody was blocked by using skimmed milk (3.0% in PBS). Cells were incubated overnight at 4 °C with anti-ICAM-1 mAb or anti-VCAM-1 antibody, diluted in blocking buffer, the cells were further washed with PBS and incubated with peroxidase-conjugated goat anti-mouse secondary Abs. After washings, cells were exposed to the peroxidase substrate (o-phenylenediamine dihydrochloride 40 mg/100 ml in citrate phosphate buffer, pH 4.5). Reaction was stopped by the addition of 2 N sulfuric acid and absorbance at 490 nm was measured using microplate reader (Spectramax 190, Molecular Devices, USA).
Neutrophil isolation
Neutrophils were isolated from peripheral blood of healthy individuals as described earlier.45 Briefly, venous blood was collected from healthy individuals in heparin solution (20 U ml−1) and erythrocytes were removed by sedimentation against 6% dextran solution. Plasma, rich in white blood cells, was layered over Ficoll–Hypaque solution, followed by centrifugation (300 g for 20 min, 20 °C). The top saline layer and the Ficoll–Hypaque layer were aspirated leaving neutrophils/RBC pellet. The residual red blood cells were removed by hypotonic lysis. Isolated cells were washed with PBS and re-suspended in PBS containing 5 mM glucose, 1 mM CaCl2, and 1 mM MgCl2 at a final concentration of 6 × 105 cells per ml.
Cell adhesion assay
This was performed as described earlier.45 Briefly, the endothelial cells plated in 96-well culture plates were incubated with or without compound 11 at desired concentrations for 2 h, followed by induction with TNF-α (10 ng ml−1) for 6 h. Endothelial monolayers were washed with PBS and neutrophils (6 × 104 per well) were added over it and were allowed to adhere for 1 h at 37 °C. The non-adherent neutrophils were washed with PBS and neutrophils bound to endothelial cells were assayed by adding a substrate solution consisting of o-phenylenediamine dihydrochloride (40 mg/100 ml in citrate phosphate buffer, pH 4.5), 0.1% cetrimethyl ammonium bromide, and 3-amino-1,2,4 triazole (1 mM). The absorbance was read at 490 nm using an automated microplate reader (Model 680, Bio-Rad, USA).
Pharmacokinetics
The compound 11 was administered orally (20 mg kg−1) or through intravenous route (10 mg kg−1 i.v.) as a fine suspension in 1% gum acacia. Blood samples were drawn from retro-orbital plexus at designated times in pre-labeled heparinised tubes and centrifuged (3500 rpm × 10 minutes) to obtain the plasma. Blood samples were collected at 0, 5, 15 and 30 min, and 1, 2, 4, 6, 8, 16 and 24 h post-administration after oral/i.v. route.
Aliquots of plasma were mixed with acetonitrile (1:2), swirled for two minutes (2500 rpm) and centrifuged at 5000 rpm for 10 min. The organic layer was collected and evaporated to dryness using solvent evaporator (Model: SPD 111V, Thermo Electron Corporation, MA). The dried samples were reconstituted in mobile phase for HPLC analysis. In a similar manner, samples were also prepared from aliquots of plasma collected from untreated animals which were spiked with compound 11 (0.25–10 μg ml−1). These were used to draw calibration curves which were found to be linear (r2 = 0.9998). Reproducibility of the method was defined by both intra-, and inter-day variance. The retention time of compound 11 was 10.688 min, at which no other interfering peak was observed. The lower limit of detection (LOD) was 11 ng ml−1; limit of quantitation (LOQ) was 32 ng ml−1; recovery was 90 ± 2%. HPLC determinations were done on Shimadzu chromatograph (Model: LC-10 Atvp equipped with a diode array detector). Compound 7 was determined at 276 nm using RP-18 column (5 μm × 25 cm). Mobile phase consisted of acetonitrile: water (55:45) with a flow rate of 1.0 ml min−1. A concentration–time curve for compound 11 was established and pharmacokinetic parameters AUC, Cmax, and tmax were determined by a non-compartmental analysis using TOPFIT software package. Absolute bioavailability was determined using the following formula: AUCoral × dosei.v./AUCi.v. × doseoral.
Cytotoxicity assay
Basal cytotoxicity of compound 11 was performed to measure IC50. Balb/c 3T3 Neutral Red Dye Uptake (NRU) cytotoxicity assay procedure was adopted.46 In brief, 3T3 cells were grown in 96 well microtiter plates and exposed to compound 11, vehicle control or untreated vehicle control. After 48 hours incubation, the test material was removed, cells rinsed twice with 250 μl of pre-warmed Dulbecco's Phosphate Buffered Saline (D-PBS) and thereafter incubated with Neutral Red (NR) medium at 37 °C, 90% humidity and 5% CO2/air for ∼3 h. Wells showing NR crystalizations were rejected. After incubation, the NR medium was removed, and cells rinsed with 250 μl pre-warmed D-PBS and added 100 μl NR desorb (EtOH/acetic acid) solution to all wells, including blanks. NR was extracted from the cells by placing the microtiter plate on a microtiter plate shaker for 30 minutes and absorption measured at 540 nm in a microtiter plate reader (spectrophotometer), using the blanks as a reference. A calculation of cell viability expressed as NRU was made for each concentration of compound 11 by using the mean NRU of 4 to 6 replicate values per test concentration. The cell viability value was compared with the mean NRU of all vehicle control values and relative cell viability was then expressed as percent of untreated vehicle control.
Statistical analysis
The data are presented as the mean ± SEM. All the experiments were repeated independently at least 3 times. Data between two groups were evaluated by Students t-test (independent) using Microcal Origin 6.0 (Microcal Software Inc, MA, USA). A level of P < 0.05 was set as the threshold for statistical significance between the control and various experimental groups.
Conclusion
We demonstrate synthesis of novel triazine-aryl-bis-indole derivatives which inhibit phosphodiesterase and stabilized cAMP in in vivo conditions in association with inhibition of adhesion molecules. These compounds might have therapeutic potential for the treatment of various inflammatory diseases.
Acknowledgements
This work is supported by grants (NWP 033 and BSC 0116) from Council of Scientific and Industrial Research, New Delhi, India.
References
- K. S. Yeung, Drug Discovery Today, 2009, 14, 812–813 CrossRef PubMed
 .
. - M. D. Houslay, P. Schafer and K. Y. J. Zhang, Drug Discovery Today, 2005, 10, 1503–1519 CrossRef CAS .
- V. Dal Piaz and M. P. Giovannoni, Eur. J. Med. Chem., 2000, 35, 463–480 CrossRef CAS .
- N. A. Molfino, Respiration, 2005, 72, 105–112 CrossRef PubMed .
- M. Conti and J. Beavo, Annu. Rev. Biochem., 2007, 76, 481–511 CrossRef CAS PubMed .
- M. M. Teixeira, R. W. Gristwood, N. Cooper and P. G. Hellewell, Trends Pharmacol. Sci., 1997, 18, 164–170 CrossRef CAS .
- T. J. Torphy, Am. J. Respir. Crit. Care Med., 1998, 157, 351–370 CrossRef CAS PubMed .
- M. J. Sanz, J. Cortijo and E. J. Morcillo, Pharmacol. Ther., 2005, 106, 269–297 CrossRef CAS PubMed .
- W. M. Brown, Int. J. Chronic Obstruct. Pulm. Dis., 2007, 2, 517–533 CAS .
- K. Higuchi and T. Kawasaki, Nat. Prod. Rep., 2007, 24, 843–868 RSC .
- S. E. O. Connor and J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC .
- S. Olgen, A. Kaessler, D. Nebioglu and J. Joachim, Chem. Biol. Drug Des., 2007, 70, 547–551 CAS .
- B. P. Smart, R. C. Oslund, L. A. Walsh and M. H. Gelb, J. Med. Chem., 2006, 49, 2858–2860 CrossRef CAS PubMed .
- C. R. Hopkins, M. Czekaj, S. S. Kaye, Z. Gao, J. Pribish, H. Pauls, G. Liang, K. Sides, D. Cramer, J. Cairns, Y. Luo, H. K. Lim, R. Vaz, S. Rebello, S. Maignan, A. Dupuy, M. Mathieu and J. Levell, Bioorg. Med. Chem. Lett., 2005, 15, 2734–2737 CrossRef CAS PubMed .
- F. Leroux, B. J. van keulen, J. Daliers, N. Pommery and J. P. Henichart, Bioorg. Med. Chem. Lett., 1999, 7, 509–516 CrossRef CAS .
- A. Makhlouf, A. Kshirsagar and C. Niederberger, Int. J. Impotence Res., 2006, 18, 501–509 CrossRef CAS PubMed .
- V. Boswell-Smith, D. Spina and C. P. Page, Pharmacology, 2006, 147, S252–S257 CAS .
- N. Kaushik, A. Pankaj, K. Naresh, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed .
- A. D. Brown, M. E. Bunnage, P. A. Glossop, M. Holbrook, R. D. Jones, C. Lane, R. A. Lewthwaite, S. Mantell, C. Perros-Huguet, D. A. Price and R. Webster, Bioorg. Med. Chem. Lett., 2007, 17, 6188–6191 CrossRef CAS PubMed .
- S. Zhang, Z. Li, X. Wu, Q. Huang, H. M. Shen and C. N. Ong, Mol. Cancer Ther., 2006, 5, 3285–3293 CrossRef CAS PubMed .
- S. Safe, S. Papineni and S. Chintharpalli, Cancer Lett., 2008, 269, 326–338 CrossRef CAS PubMed .
- F. Akahoshi, S. Takeda, T. Okada, M. Kajii, H. Nishimura, M. Sugiura, Y. Inoue, C. Fukaya, Y. Naito, T. Imagawa and N. Nakamura, J. Med. Chem., 1998, 41, 2985–2993 CrossRef CAS PubMed .
- C. Hulme and K. Moriarty, Bioorg. Med. Chem. Lett., 1998, 8, 1867–1872 CrossRef CAS .
- P. Fireman, Allergy Asthma Proc., 2003, 24, 79–83 CAS .
- K. F. Rabe, H. Magnussen and G. Dent, Eur. Respir. J., 1995, 8, 637–642 CAS .
- E. Lopez, P. H. Jarreau, E. Zana, M. L. Franco-Montoya, T. Schmitz, D. Evain-Brion, J. Bourbon, C. Delacourt and C. Méhats, Dev. Dyn., 2010, 239, 2470–2478 CrossRef CAS PubMed .
- T. Muller, P. Engels and J. R. Fozard, Trends Pharmacol. Sci., 1996, 17, 294–298 CrossRef CAS .
- P. Reid, Curr. Opin. Invest. Drugs, 2002, 3, 1165–1170 CAS .
- P. Christie, Drugs Today, 2005, 41, 667–675 CrossRef CAS PubMed .
- C. Cowan, Issues Emerg. Health Tech., 2005, 74, 1–4 Search PubMed .
- S. A. Antoniu, Curr. Opin. Invest. Drugs, 2006, 7, 412–417 CAS .
- S. Minguet, M. Huber, L. Rosenkranz, W. W. Schamel, M. Reth and T. Brummer, Eur. J. Immunol., 2005, 35, 31–41 CrossRef CAS PubMed .
- J. A. Nogueira-Machado, F. C. Lima e Silva, E. P. Cunha, M. R. Calsolari, D. C. Costa, C. S. Perilo, B. C. Horta, I. C. Ferreira and M. M. Chaves, Diabetes Metab., 2006, 32, 331–335 CAS .
- M. Filipović and S. Cekić, Biol. Med., 2001, 8, 6–10 Search PubMed .
- L. A. Stanciu and R. Djukanovic, Eur. Respir. J., 1998, 11, 949–957 CrossRef CAS .
- Y. Delneste, P. Jeannin, P. Gosset, P. Lassalle, E. Cardot, I. Tillie-Leblond, M. Joseph, J. Pestel and A. B. Tonnel, Clin. Exp. Immunol., 1995, 101, 164–171 CrossRef CAS PubMed .
- R. Louis, J. Shute, S. Biagi, L. Stanciu, F. Marrelli, H. Tenor, R. Hidi and R. Djukanović, Am. J. Respir. Crit. Care Med., 1997, 155, 466–472 CrossRef CAS PubMed .
- K. A. Kim, J. S. Lee, H. J. Park, J. W. Kim, C. J. Kim, I. S. Shim, N. J. Kim, S. M. Han and S. Lim, Life Sci., 2004, 74, 2769–2779 CrossRef CAS PubMed .
- L. C. Chiang, W. Chiang, M. Y. Chang, L. T. Ng and C. C. Lin, Am. J. Chin. Med., 2003, 31, 37–46 CrossRef CAS PubMed .
- J. R. Gurr, A. S. Wang, C. H. Chen and K. Y. Jan, Toxicology, 2005, 213, 66–73 CrossRef CAS PubMed .
- M. Baeeri, A. Foroumadi, M. Motamedi, A. Yahya-Meymandi, L. Firoozpour, S. N. Ostad, A. Shafiee, S. Saeid and M. Abdollahi, Chem. Biol. Drug Des., 2011, 78, 438–444 CAS .
- W. C. Ko, C. M. Shih, Y. H. Lai, J. H. Chen and H. L. Huang, Biochem. Pharmacol., 2004, 68, 2087–2094 CrossRef CAS PubMed .
- N. Seguy, H. F. Hildebrand and J. M. Haguenoer, Toxicol. Lett., 1994, 70, 23–32 CrossRef CAS .
- I. S. Choi, B. S. Kim, K. S. Cho, J. C. Park, M. H. Jang, M. C. Shin, S. B. Jung, J. H. Chung and C. J. Kim, Toxicol. Lett., 2002, 132, 47–55 CrossRef CAS .
- S. Kumar, V. Singhal, R. Roshan, A. Sharma, G. W. Rembhotkar and B. Ghosh, Eur. J. Pharmacol., 2007, 575, 177–186 CrossRef CAS PubMed .
- E. Borenfreund and J. A. Puerner, Toxicol. Lett., 1985, 24, 119–124 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra11495k |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 









