
Properties and structures of β-glucuronidases with different transformation types of glycyrrhizin†
 *ab
*ab
Abstract
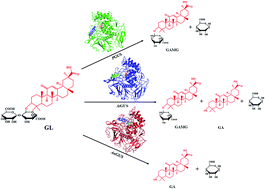
β-Glucuronidase (GUS) has been widely used to hydrolyze β-linked glucuronides to generate various valuable derivatives. In this study, the GUS from three fungi (PGUS from P. purpurogenum Li-3, AtGUS from A. terreus Li-20 and AuGUS from A. ustus Li-62) with significantly different types of glycyrrhizin (GL) hydrolysis were comparatively investigated. The Km values of PGUS, AtGUS and AuGUS were 0.328, 3.61 and 0.429 mM respectively. These results indicated that AtGUS showed the lowest affinity for GL among the three kinds of GUS, while PGUS and AuGUS had a similar affinity for GL. Nevertheless, the Vmax/Km values showed that AuGUS had the highest catalytic efficiency for GL hydrolysis, so it was supposed to be an efficient biocatalysis for GL biotransformation. The sequence properties analysis demonstrated that the three GUS had some special different sequence characteristics, but that is not the key reason for the discrepancy in catalytic type. The homologous modeling analysis indicated that various GL transformation types of PGUS, AtGUS and AuGUS were likely caused by the different positions of bacterial loops surrounding their aglycone binding pocket. These results can not only help us to better understand the catalytic diversity of GUS, but also give us an important guide to redesign the catalytic diversity of GUS.
 Please wait while we load your content...
Please wait while we load your content...

