
Compressed sodalite-like MgH6 as a potential high-temperature superconductor
Abstract
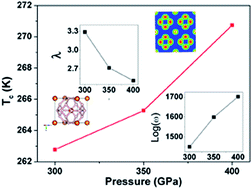
Recently, an experimental work reported a very high Tc of ∼190 K in hydrogen sulphide (H2S) at 200 GPa. The search for new superconductors with high superconducting critical temperatures in hydrogen-dominated materials has attracted significant attention. Here we predict a candidate phase of MgH6 with a sodalite-like framework in conjunction with first-principles electronic structure calculations. The calculated formation enthalpy suggests that it is thermodynamically stable above 263 GPa relative to MgH2 and solid hydrogen (H2). Moreover, the absence of imaginary frequency in phonon calculations implies that this MgH6 structure is dynamically stable. Furthermore, our electron–phonon coupling calculation based on BCS theory indicates that this MgH6 phase is a conventional superconductor with a high superconducting critical temperature of ∼260 K under high pressure, which is even higher than that of the recently reported compressed H2S. The present results offer insight in understanding and designing new high-temperature superconductors.
 Please wait while we load your content...
Please wait while we load your content...

