DOI:
10.1039/C5RA11448A
(Paper)
RSC Adv., 2015,
5, 60467-60481
Novel contribution to the simultaneous analysis of certain hypoglycemic drugs in the presence of their impurities and degradation products utilizing UPLC-MS/MS
Received
15th June 2015
, Accepted 3rd July 2015
First published on 6th July 2015
Abstract
A novel ultra-performance liquid chromatography coupled to tandem mass spectrometry (UPLC-MS/MS) method was established for simultaneous determination of three hypoglycemic drugs namely; sitagliptin (STG), vildagliptin (VLG) and metformin (MET) in the presence of their degradation products and STG related impurities. Chromatographic separation was accomplished on a Hypersil gold 50 mm × 2.1 mm (1.9 μm) column, using acetonitrile and 0.2% formic acid aqueous solution as the mobile phase with a gradient elution. Electrospray ionization (ESI) source was operated in the positive ion mode. The selected reaction monitoring (SRM) mode on a triple quadrupole mass spectrometer was used to quantify the drugs utilizing the transitions of 408.12 → 235.24 (m/z), 304.33 → 154.32 (m/z), 130.12 → 71.32 (m/z) and 255.75 → 166.15 (m/z), for STG, VLG, MET and diphenhydramine (IS), respectively. The method has displayed a lower limit of detection of 1.50 ng mL−1, 1.50 ng mL−1 and 3.00 ng mL−1 for STG, VLG and MET, respectively. The drugs were subjected to forced degradation where it was concluded that STG, VLG and MET were highly susceptible for alkaline stress conditions. In addition, the study of the degradation kinetics of the drugs has proved that the degradation follows a pseudo-first-order reaction. The proposed method was effectively applied for the analysis of laboratory prepared mixtures as well as combined pharmaceutical formulations. No significant difference was found regarding accuracy and precision upon statistical comparison of the obtained results with those of the reported method. Validation was conducted in compliance with the ICH guidelines proving the method to be selective, linear, precise and accurate. The simplicity and sensitivity of this method allows its use in the quality control of the cited drugs.
1. Introduction
Sitagliptin (STG), and vildagliptin (VLG), Table 1, are relatively new oral antihyperglycemic drugs that belong to the dipeptidyl-peptidase-4 inhibitor (DPP-4) class which functions through stimulating glucose-dependent insulin release.1–3 DPP-4 inhibitors represent a novel therapeutic approach for treatment of type-II diabetes; that improved glycemic control by preventing glucagon-like peptide-1 (GLP-1) and glucose dependent insulinotropic polypeptide (GIP) degradation. These intestinal peptides, also known as incretins, are postprandially secreted and lead to a rise in insulin secretion.4 Metformin (MET), Table 1, is a biguanide drug that stimulates glycolysis in peripheral tissues.5 It functions by lowering blood sugar and allows the body to use insulin more efficiently. Lately, the combination of STG and MET has showed effectiveness in controlling the metabolic syndrome and has caused significant weight loss, reversal of insulin resistance, islet and adipocyte hypertrophy and alleviated hepatic steatosis.6 Moreover, the combination of VLG and MET is specifically used for patients with type-II-diabetes to help control their glycaemia levels. It is typically taken when other diabetic medications (such as pioglitazone or glicazine) have proven unsuccessful. Hitherto, STG and VLG are not official in any of the pharmacopoeias as they are considered as relatively new products but MET is official in British Pharmacopoeia (BP)7 and United State Pharmacopoeia.8
Table 1 Chemical structures and chemical names of sitagliptin, metformin, vildagliptin, diphenhydramine (IS) and sitagliptin impurities
| Name |
Structure |
IUPAC name |
| Sitagliptin |
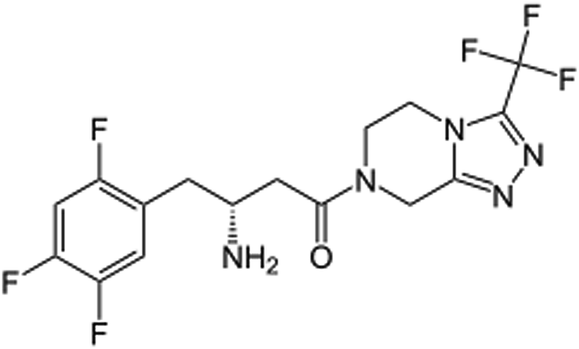
 |
(R)-4-Oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine |
| Metformin |
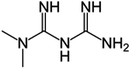 |
3-(Diaminomethylidene)-1,1-dimethylguanidine |
| Vildagliptin |
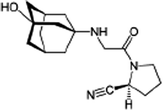 |
(2S)-1-[2-[(3-hydroxy-1-adamantyl)amino]acetyl]pyrrolidine-2-carbonitrile |
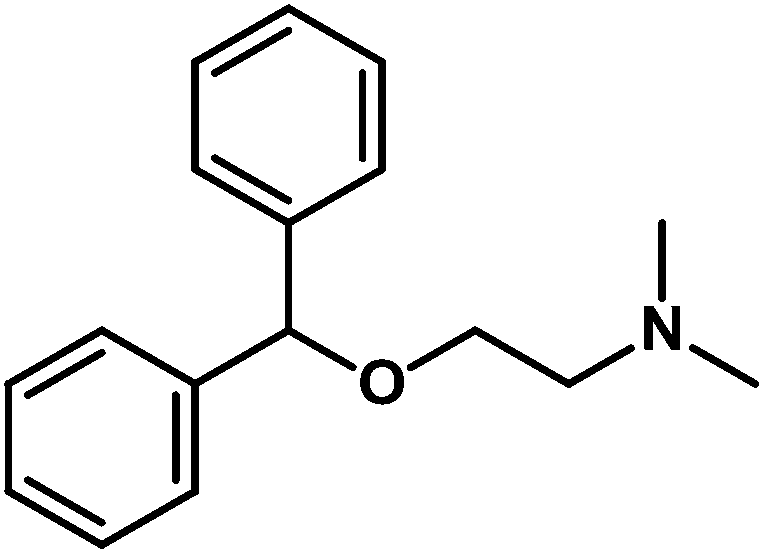
| Diphenhydramine (IS) |
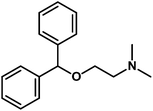 |
2-Benzhydryloxy-N,N-dimethylethanamine |
| Sitagliptin impurity 1 |
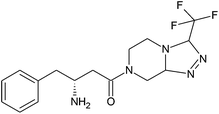 |
(R)-4-Oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-butan-2-amine |
| Sitagliptin impurity 2 |
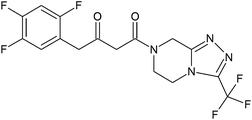 |
(2Z)-4-Oxo-4-[3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-one |
Literature survey has revealed several methods for estimation of MET and STG in pharmaceutical formulations or in biological fluids. These methods have included spectrophotometry and spectrofluorimetry,9 HPLC and stability-indicating HPLC methods.10–16 Considering the aforementioned stability indicating studies, STG and MET were determined in presence of their degradation products by HPLC-UV in concentration ranges of 10–30 ppm and 100–300 ppm, respectively as performed by Kumar et al.13 R. I. El-Bagary et al.,11 have also utilized HPLC-UV for quantification of STG and MET in presence of STG alkaline degradation product in the range of 5–160 μg mL−1 and 25–800 μg mL−1, respectively. Simultaneous estimation of STG, MET and their impurities was carried out by a stability indicating method manifesting a good separation using HPLC-UV, where STG and MET were estimated in concentration range of 10–150 μg mL−1 and 50–450 μg mL−1, respectively.12 In addition to the previously mentioned methods, a UPLC-UV method for simultaneous assay of STG (in the range 25–75 μg mL−1) and MET (in the range 250–750 μg mL−1) was reported by Malleswararao et al.,15 where the authors have conducted forced degradation to demonstrate the selectivity of the method. This method has revealed four impurities from basic degradation and three impurities from oxidative stress. Different LC-MS methods were also developed,17,18 where S. L. Bonde et al.,18 have simultaneously determined the drugs in the range of 5–800 ng mL−1 for STG and 25–3000 ng mL−1 for MET. However none of these LC-MS methods was stability-indicating. In addition capillary electrophoresis was also reported for the determination of STG in combination with MET.19
Regarding the simultaneous determination of VLG and MET either in pharmaceutical dosage forms or in biological fluids, Abdel-Ghany et al.20 have recently established five new spectrophotometric methods for the analysis of VLG and MET in binary mixture. In addition, several HPLC-UV methods were performed.16,21–24 However, LC methods based on UV and DAD detectors show moderate sensitivity and selectivity for complex matrices such as plasma. Thus all of these methods21–24 have revealed microgram linearity ranges for both drugs. One of the previously mentioned HPLC-UV methods was extended to include stability studies on both VLG and MET and has concluded that the drugs were stable under their applied conditions.21 Regarding mass spectrometry; R. Pontarolo et al.25 have developed and validated a HILIC-MS/MS method for simultaneous determination of VLG and MET over the ranges of 5–500 ng mL−1 for each drug. Recently, C. P. Uber et al.26 have performed an HPLC-MS/MS method for simultaneous determination of VLG (5–150 ng mL−1), MET (250–2000 ng mL−1) and MET-related compounds in tablets. However, neither of the latter two methods was a stability indicating one.
To the best of our knowledge, no quantitative UPLC-MS/MS method was reported concerning the simultaneous analysis of STG, VLG and MET in presence of the three drugs' degradation products or impurities. Thus, our main aim was to develop a validated, sensitive, selective and rapid UPLC-MS/MS method for their simultaneous analysis. In addition to perform a comparative study between recently developed HPLC-MS/MS methods for the determination of binary combinations of these drugs regarding the chromatographic conditions to shed the light on the advantages offered by our proposed method. Moreover, to introduce a comparison between the differences of the stress conditions applied on the drugs either by our method or other reported ones to show how the applied conditions affect the produced degradation products and the percentage degradation. Finally was to study the degradation kinetics of the drugs.
2. Experimental
2.1. Instruments
The analysis was achieved using a TSQ Quantum Access MAX triple stage quadrupole mass spectrometer, Thermo Scientific, New York, USA, equipped with an electrospray ionization (ESI) source. The control of the LC-MS/MS system, collection and analysis of the data was performed utilizing Xcalibur software version 2.2. Chromatography was carried on Accela U-HPLC system which was composed of Accela 1250 quaternary pump and Accela open autosampler, New York, USA (operated at 25 °C).
2.2. Chemicals and reagents
Sitagliptin phosphate monohydrate reference standard material was kindly supplied by Merck Sharp and Dohme Co. (Cairo, Egypt). Its purity was found to be 99.76% according to a reported method.16 Vildagliptin was supplied from Novartis Europharm Ltd. Co. (London, U.K.). Its purity was found to be 99.64% according to a reported method.16 Metformin hydrochloride was kindly supplied by Chemical Industries Development (CID) Co. (Giza, Egypt). Its purity was found to be 100.04% according to a reported method.16 Diphenhydramine (IS) was kindly supplied by Sigma Pharmaceutical Industries, Steinheim, Germany. Janumet® tablets nominally containing 50 mg of STG and 1000 mg of MET per tablet B.N. N17560 (Merck Sharp and Dohme Co. Cairo, Egypt) was purchased from local market. Galvus Met® tablets nominally containing 50 mg of VLG and 500 mg of MET per tablet B.N. W0011, Novartis Europharm Ltd. Co. (London, U.K.) was purchased from local market. Sitagliptin impurity 1 (IMP1) and sitagliptin impurity 2 (IMP2) are shown in Table 1, they were kindly supplied by TLC PharmaChem Inc (Ontario, Canada). Human plasma was purchased from Vaccera (Giza, Egypt), kept frozen until used after gentle thawing.
All chemicals used throughout the work were of analytical grade and solvents were of HPLC grade. Methanol was purchased from Fisher Scientific UK Ltd, Loughborough, UK. Acetonitrile and sodium hydroxide were purchased from Merck, Darmstadt, Germany. Hydrochloric acid 30% and hydrogen peroxide 30% were purchased from Prolabo, Pennsylvania, USA. Deionized water (Purelab flex, ELGA, Buckinghamshire, UK) was used.
2.3. Chromatographic and mass spectrometric conditions
Chromatographic separation was accomplished on Hypersil-Gold column (C18-bonded ultrapure silica based column) 50 mm × 2.1 mm (1.9 μm) from Thermo Scientific, New York, USA. Gradient elution was achieved using the binary mobile phase consisting of 0.2% formic acid aqueous solution (A) and acetonitrile (B) using a flow rate of 250 μL min−1, where elution was performed at room temperature. A gradient program was conducted as follows: 10% B from 0–0.2 min, ramp to 90% B from 0.2–0.7 min, hold at 90% B till 2 min, back to 10% B from 2.0–3.0 min. The injection volume was 10 μL and the total run time for each sample was 3 min.
The mass spectrometric detection method was carried out in the positive-ion utilizing electrospray ionization (ESI) and selected reaction monitoring (SRM) mode. The optimized parameters are: auxiliary gas of 2 psi, sheath gas of 15 psi, capillary temperature of 270 °C, turbo ion spray temperature of 400 °C, ion spray voltage of 3500 V and capillary offset of 35 V. The quadrupole mass spectrometer was operated at the SRM mode, monitoring the transition of molecular ions to the product ions for STG m/z 408.12 → 235.24, VLG 304.33 → 154.32, MET 130.12 → 71.32 and IS 255.75 → 166.16 using collision energy of 25 eV for each.
2.4. Procedures
2.4.1. Preparation of calibration standards. Stock solutions of 0.1 mg mL−1 for each of STG, VLG, MET and IS were separately prepared in methanol. Working standard solutions were prepared by diluting the corresponding stock solutions with methanol. The stock and working solutions were preserved at 4 °C and discarded within 30 days.
2.4.2. Construction of calibration curves. The bulk calibration curves were constructed by preparing six standard solutions of each drug in the concentration ranges of 5.0–100.0 ng mL−1 for STG, 5.0–500.0 ng mL−1 for VLG and 10.0–400.0 ng mL−1 for MET followed by the addition of IS (40 ng mL−1 final concentration) on every standard solution. The solutions were filtered through a nylon membrane filter (0.45 μm) followed by the injection of 10 μL aliquots of each solution onto the LC-MS system. The calibration curve was then constructed for each drug by plotting of the peak area ratios of each drug to IS obtained against the corresponding concentrations.
2.4.3. Assay of laboratory prepared mixtures. The working solutions of STG, VLG and MET were mixed in variable ratios to prepare different binary and ternary laboratory prepared mixtures then 40.0 ng mL−1 (final concentration) of IS was added. The solutions were filtered through a nylon membrane filter (0.45 μm) followed by the injection of 10 μL aliquots of each mixture onto the LC-MS system. The percentage recoveries were calculated by means of the corresponding regression equations or referring to the calibration graphs.
2.4.4. Application to pharmaceutical preparation. Ten tablets of Janumet® and ten tablets of Galvus Met® were separately finely powdered; accurate amounts of the powdered tablets equivalent to 20.0 mg of MET and 1.0 mg of STG (for Janumet® tablets) and equivalent to 10.0 mg of MET and 1.0 mg of VLG (for Galvus Met® tablets), were transferred into 100 mL volumetric flask. The two solutions were separately sonicated for 20 min with 30 mL methanol. Then, the volume was completed with methanol and filtered to prepare a stock solution with the concentration of 0.2 mg mL−1 of MET and 0.01 mg mL−1 of STG (for Janumet® tablets) and 0.1 mg mL−1 of MET and 0.01 mg mL−1 of VLG (for Galvus Met® tablets). The procedure was continued as stated under calibration curves “2.4.2. Construction of calibration curves”. The percentage recoveries were calculated by referring to the calibration graphs or using the corresponding regression equations.
2.4.5. Application of standard addition technique. The standard addition technique was applied to check the validity of the proposed LC-MS/MS method. Five portions of the previously powdered tablets each claimed to contain 10.0 mg MET and 0.5 mg STG (for Janumet® tablets) and 10.0 mg MET and 1.0 mg VLG (for Galvus Met® tablets) were accurately weighed and mixed with different amounts of the pure drugs, in five 100 mL volumetric flasks and sonicated for 20 min with 30 mL methanol for each. Then the volume was completed with methanol followed by filtration to prepare stock solutions. Consequently, further dilution with methanol was done to obtain the final concentration in nanogram range, followed by the addition of IS (40 ng mL−1 final concentration), and then the samples were ready to be injected onto LC-MS system. The percentage recoveries were calculated by referring to the calibration graphs or using the corresponding regression equations.
2.4.6. Forced degradation conditions. To check the applicability of the proposed UPLC-MS/MS method as a stability indicating method, forced degradation study under different stress conditions was carried out in which STG, VLG and MET were subjected to the following conditions: for alkaline and acidic hydrolysis; accurately weighed amounts 10.0 mg of each drug was treated separately with 20 mL of 1.0 N NaOH (alkaline) or 20 mL of 1.0 N HCl (acidic). The solution was heated in thermostatic water bath at 100 °C for different time intervals 5, 10, 15, 20, 30, 60 and 120 minutes and then neutralized with pre-calculated volumes of 1.0 N HCl (alkaline) or 1.0 N NaOH (acidic). For thermal degradation; an accurately weighed amount 10.0 mg of each drug was separately treated with 20 mL of deionized water and then heated for time intervals 5, 10, 15, 20, 30, 60 and 120 minutes at 100 °C followed by cooling. For oxidative degradation; an accurately weighed amount 10.0 mg of each drug was separately treated with 20 mL of 0.6% hydrogen peroxide (w/v) and left for different time intervals 5, 10, 15, 20, 30, 60 and 120 minutes at room temperature followed by sonication. The above prepared solutions were completed with methanol to obtain stock degradates solutions equivalent to 0.1 mg mL−1. Consequently, further dilution with methanol was done to obtain the final concentration in nanogram range ready to be injected onto LC-MS system after filtration through a 0.45 μm nylon membrane filter.
2.5. Method validation
The validation of the proposed method has included linearity, accuracy, precision, specificity, limits of detection and quantification and robustness according to the procedures recommended by ICH Q2 (R1).27
3. Results and discussion
The main aim of this work was to establish and validate a kinetic stability indicating UPLC-MS/MS method for the simultaneous estimation of STG, VLG and MET and their related impurities in their bulk powders and combined dosage forms. Concerning the stability indicating study; the current International Conference on Harmonization (ICH) guidelines necessitate that stability indicating assay methods should be performed and validated after applying stress testing on the drug under different conditions, including hydrolysis at various pHs, oxidation and thermal degradation.28
3.1. Method development
Aiming for the optimum detection of STG, VLG, MET, IS, related impurities and different degradates; both the mass spectrometric parameters and the chromatographic conditions were thoroughly studied. Regarding the mass spectrometric parameters, the precursor ions and product ions were adjusted by infusion of 1.00 μg mL−1 neat solutions into the mass spectrometer which was operated in positive polarity mode using electrospray ionization technique. The ions were scanned in a mass range of 100–500 m/z. As STG, VLG and MET are basic in nature, thus, having the ability to accept protons, so the intensity of their precursor ions and product ions was found to be ideal in the positive mode. The protonated molecular ions [M + H]+ 408.12 for STG, 304.33 for VLG, 130.12 for MET, 255.75 for IS, 356.3 for IMP1 and 407.12 for IMP2 were all observed on the full scan mass spectra. Additionally the [M + H]+ of the most abundant degradation products 215.25 for STG acid degradation, 193.35 for STG alkaline degradation, 154.32 and 168.37 for VLG degradation and 71.32 and 60.36 for MET degradation were also detected.
The collision energy employed in Q2 has resulted in the production of characteristic ions. Upon utilization of sufficient collision activated dissociation gas and collision energy, the following MS/MS transitions were carefully chosen 408.12 → 235.24, 304.33 → 154.32, 130.12 → 71.32, 255.75 → 166.16, 356.13 → 256.38 and 407.12 → 193.33 for STG, VLG, MET, IS, IMP1 and IMP2, respectively, as these represented the most abundant products ions (Fig. 1). Moreover, as both the capillary temperature and sheath gas flow play an important role in altering the sensitivity, thus, they also should be optimized. Adjustment of capillary temperature at 270 °C and sheath gas at 15 psi, has improved the intensity of the analytes. On the other hand, slight changes in ion spray voltage showed no obvious effect on the signal intensity and it was maintained at 3500 V.
 |
| | Fig. 1 Product ion spectra of [M + H]+ of: (a) MET, (b) VLG, (c) IS, (d) STG, (e) IMP1 and (f) IMP2 with the proposed structures of the main product ions. | |
Concerning the optimization of the chromatographic conditions; the proper choice of the mobile phase as well as the selection of column was of great importance in order to attain the best chromatographic separation with the required response. Chromatographic analysis of the drugs, IS, most abundant degradates and impurities was firstly tried using various combination of acetonitrile, methanol and 0.2% formic acid aqueous solution with varying ratios. However, methanol-containing mobile phase has resulted in bad separation as well as bad resolution of peaks for all the compounds. Thus, acetonitrile and 0.2% formic acid were the mobile phase of choice where a gradient system was developed. The ratio of acetonitrile was gradually increased from 10% till 90% within 0.7 min, then maintained at the final ratio for 1.3 min, finally, the gradient was returned to the starting conditions; (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 v/v) acetonitrile:formic acid (0.2% aqueous solution) in 1 min, thus, the total run time was 3 min. Formic acid solution has assisted in accomplishing good response for MS detection functioning in the positive mode. The described gradient system produced the best peak shape, where, the analytes were protonated and well separated within 3 min at a flow rate of 250 μL min−1 (Fig. 2 & 3). In addition, two different columns Hypersil-Gold (C18-bonded ultrapure silica based column) and Bio Basic-8 were investigated for best chromatographic performance. Hypersil-Gold (50.0 mm × 2.1 mm, 1.9 μm particle size) column which was maintained at room temperature gave better peak shape and response even at LOQ levels for the analytes.
:90 v/v) acetonitrile:formic acid (0.2% aqueous solution) in 1 min, thus, the total run time was 3 min. Formic acid solution has assisted in accomplishing good response for MS detection functioning in the positive mode. The described gradient system produced the best peak shape, where, the analytes were protonated and well separated within 3 min at a flow rate of 250 μL min−1 (Fig. 2 & 3). In addition, two different columns Hypersil-Gold (C18-bonded ultrapure silica based column) and Bio Basic-8 were investigated for best chromatographic performance. Hypersil-Gold (50.0 mm × 2.1 mm, 1.9 μm particle size) column which was maintained at room temperature gave better peak shape and response even at LOQ levels for the analytes.
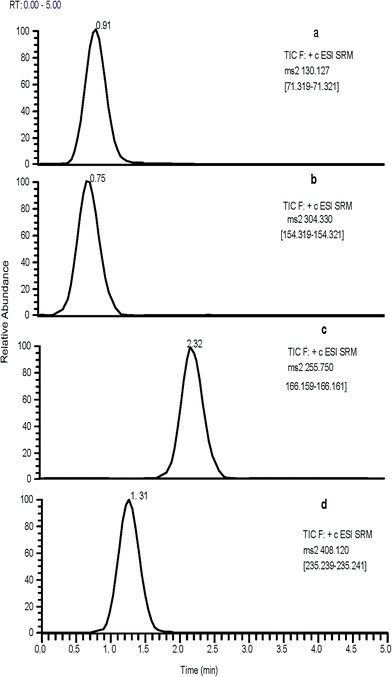 |
| | Fig. 2 Representative SRM chromatograms of: (a) MET, (b) VLG, (c) IS and (d) STG. | |
 |
| | Fig. 3 Representative Q1MS chromatograms of the main degradation products of: (a & b) VLG, (c & d) MET. (e) Acid degradation product of STG, (f) alkaline degradation product of STG, (g) SRM chromatogram of IMP1 and (h) SRM chromatogram of IMP2. | |
After optimization of both mass spectrometric and chromatographic conditions; well-defined peaks and good sensitivity were accomplished within a short run time which is not achievable using the classical HPLC.
A thorough literature survey has revealed that our proposed method is the first performed attempt for the simultaneous determination of STG, VLG and MET in presence of their related impurities and degradation products using UPLC-MS/MS. In addition it is the first stability indicating method which was extended for studying the kinetics of the degradation reactions of the three drugs. However, four HPLC-MS/MS methods were found in the literature concerned with the simultaneous determination of binary combinations of these drugs; two of which were dealing with STG and MET17,18 and the other two methods were dealing with VLG and MET.25,26 A summary of the utilized chromatographic conditions of these methods is presented in Table 2. When taking a look on Table 2; it was obvious that our proposed method has shown a lot of advantages over all the reported methods.17,18,25,26 The utilization of UPLC with the smaller column size (50.0 mm × 2.1 mm, 1.9 μm particle size) in our proposed method has resulted in very short run time (3 min) in addition to very well resolution due to the small particle size. Moreover, we have used the lowest flow rate (250 μL min−1) which has of course offered the least solvent consumption which is considered to be cost effective specifically for quality control labs. In addition we have employed the simplest mobile phase and avoided the use of buffer. Regarding sensitivity our proposed method has shown higher sensitivity compared to S. L. Bonde et al. method18 and C. P. Uber et al. method,26 specifically for the determination of MET. Our linear range was 10.0–400.0 ng mL−1, thus, the LOQ determined by our method is approximately 2.5 and 25 times lower than the reported methods,18,26 respectively.
Table 2 Comparison between the chromatographic conditions used for separation of sitagliptin and metformin or vildagliptin and metformin in different reported HPLC-MS/MS methods and our proposed method
| |
Method |
Column |
Mobile phase |
Flow rate |
Linearity range |
| STG/MET |
S. L. Bonde et al. method18 |
Hypurity C18 column (50 mm × 4.6 mm, 5 μm particle size) maintained at 40 °C |
Acetonitrile and 10 mM ammonium formate (pH 3.5) in the ratio 60:40 v/v |
1000 μL min−1 |
5–800 ng mL−1 for STG, 25–3000 ng mL−1 for MET |
| J. Martín et al. method17 |
LiChrospher® 100 RP-18 column (125 mm × 4.0 mm, 5 μm particle size) maintained at 25 °C |
Acetonitrile (containing formic acid 0.1%, v/v) and an aqueous 10 mM ammonium formate solution (containing formic acid 0.1%, v/v) |
700 μL min−1 |
Obtained by spiking tap water with STG and MET at concentrations ranging from 0.2–200 ng L−1 |
| VLG/MET |
R. Pontarolo et al. method25 |
Atlantis HILIC silica column (150 mm × 2.1 mm, 3 μm particle size) maintained at 40 °C |
20% water and 80% acetonitrile/water solution 95:5 (v/v), containing both 0.1% formic acid and 3 mM ammonium formate |
400 μL min−1 |
5–500 ng mL−1 for VLG, 5–500 ng mL−1 for MET |
| C. P. Uber et al. method26 |
C8 column (150 × 4.6 mm, 5 μm particle size) maintained at 25 °C |
Acetonitrile/water/formic acid (20:80:0.1, v/v/v) |
800 μL min−1 |
5–150 ng mL−1 for VLG, 250–2000 ng mL−1 for MET |
| STG/MET/VLG |
Proposed method |
Hypersil-gold; C18-bonded ultrapure silica based column (50.0 mm × 2.1 mm, 1.9 μm particle size) maintained at 25 °C |
Acetonitrile and 0.2% formic acid (gradient elution ranging from 10% to 90% acetonitrile) |
250 μL min−1 |
5–100 ng mL−1 for STG, 10–500 ng mL−1 for MET, 5–500 ng mL−1 for VLG |
3.2. Method validation
3.2.1. Linearity. Under the optimum chromatographic and mass spectrometric conditions STG, VLG and MET were evaluated by analyzing six different concentrations of each drug in triplicates. Linear relationship between the peak area ratios of each analyte (STG, VLG or MET)/IS and their corresponding concentrations was established. The linearity ranges were found to be 5.0–100.0 ng mL−1 for STG, 5.0–500.0 ng mL−1 for VLG and 10.0–400.0 ng mL−1 for MET using the following regression equations:
| For STG; A = 2.74 × 10−3C − 6.93 × 10−3r = 0.9999 |
| For VLG; A = 7.22 × 10−4C + 5.32 × 10−2r = 0.9998 |
| For MET; A = 6.64 × 10−4C + 3.57 × 10−4r = 0.9999 |
where A is the peak area ratio, C is the concentration in ng mL−1 and r is the correlation coefficient. The linearity of the calibration curves were validated by the high value of correlation coefficients. The standard deviations for residuals was found to be 2.44 × 10−3, 3.17 × 10−3 and 2.18 × 10−3, the standard error for the slope was 3.64 × 10−5, 7.36 × 10−6 and 7.54 × 10−6 and the standard error for the intercept was 2.35 × 10−3, 1.71 × 10−3 and 1.80 × 10−3 for STG, VLG and MET, respectively.
3.2.2. Accuracy. The accuracy of the proposed method was assessed by examining six levels of standard solutions of the studied analytes, each three times and was found to be 99.24 ± 0.70 for STG, 100.27 ± 0.83 for VLG and 100.59 ± 0.81 for MET. The proposed method results were satisfactorily compared with those of reported method.16 The results obtained from the statistical analysis29 showed no significant difference between the performances of the proposed and reference methods using student's t-test and variance ratio F-test, (Table 3).
Table 3 Accuracy and precision data obtained by the proposed LC-MS/MS method and the reported one16 for the analysis of vildagliptin, metformin and sitagliptin in pure forma,b
| Item |
Vildagliptin |
Metformin |
Sitagliptin |
| Proposed |
Reported |
Proposed |
Reported |
Proposed |
Reported |
| SD = standard deviation, % RSD = percent relative standard deviation, values in parenthesis are the theoretical values of t and F at P = 0.05,29 *average of three different determinations. The reported method16 is an HPLC method, using a cyano column (250 mm × 4.6 mm, 5 μm). Isocratic elution using a mobile phase of potassium dihydrogen phosphate buffer pH (4.6)–acetonitrile (30:70, v/v) at a flow rate of 1 mL min−1 with UV detection at 210 nm. |
| Mean* ± SD |
100.27 ± 0.83 |
99.64 ± 0.73 |
100.59 ± 0.81 |
100.04 ± 0.95 |
99.24 ± 0.70 |
99.76 ± 0.74 |
| % RSD |
0.83 |
0.73 |
0.81 |
0.95 |
0.71 |
0.75 |
| n |
6 |
4 |
6 |
4 |
6 |
4 |
| Variance |
0.69 |
0.53 |
0.66 |
0.90 |
0.49 |
0.55 |
| t-test |
1.226 (2.306) |
0.982 (2.306) |
1.11 (2.306) |
| F-test |
1.291 (9.013) |
1.351 (5.409) |
1.13 (5.409) |
|
| Precision (RSD%, n = 9) |
| Intraday |
0.79 |
|
0.99 |
|
0.61 |
|
| Inter-day |
0.99 |
1.10 |
0.82 |
3.2.3. Precision. The intraday precision was estimated through triplicate analysis of three different concentrations of the analytes within the same day. On the other hand, the interday precision was achieved through analysis of three different concentrations of the analytes on three successive days. The RSD values were less than 2% demonstrating that the method was precise. The percentage relative standard deviations were calculated as abridged in Table 3.
3.2.4. Specificity. Specificity is the capability of the analytical method to determine the analyte response when accompanied with interferences including degradation products and related substances. The proposed method was used for the determination of STG, VLG and MET in the presence of their degradation products and two STG related impurities. One of the advantages of coupling LC with MS/MS detection in the SRM mode is high specificity, since only the ions resulting from the analytes of concern are observed. The chromatograms of the samples were monitored for the detection of any extra peaks, however, no chromatographic interference from any of the excipients was observed at the retention times of the drugs. Moreover, the chromatogram of each analyte in the sample solution was completely matching with the chromatogram received from the standard solution. These results revealed the absence of interference from any ingredients in the dosage forms and therefore approve the specificity of the proposed method.
3.2.5. Limit of detection (LOD) and limit of quantification (LOQ). The LOD was taken as the amount for which the S/N ratio was 3:1, while, the LOQ was taken as the amount for which the S/N ratio was 10:1. LOD was 1.50, 1.50 and 3.00 ng mL−1 and LOQ was 5.00, 5.00 and 10.00 ng mL−1, for STG, VLG and MET, respectively.
3.2.6. Robustness of the method. The robustness of the method was verified by the uniformity of the peak area ratios of the analytes/IS with the intentional minor changes performed in the mass spectrometric parameters and chromatographic conditions, e.g. sheath gas flow (±5 psi), the capillary temperature or turbo ion spray temperature (±5 °C), collision energy (±5 V) and the flow rate (±10 μL).
3.3. Application of the method for analysis of laboratory prepared mixture and dosage form
The proposed method was effectively applied for the analysis of laboratory prepared binary and ternary mixtures of STG, VLG and MET in which the drugs were mixed in different ratios. The average percent recoveries were established on the average of triplicate determinations (Table 4). Additionally, Janumet® tablets and Galvus Met® tablets were analyzed in order to show the capability of the proposed LC-MS method for quality control and routine analysis of the studied drugs in their dosage forms. The concentrations of the drugs were calculated referring to the corresponding regression equation. Standard addition technique was also applied so as to study the interference of commonly used tablet excipients. The mean percentage recoveries as well as the standard deviation obtained from the standard addition method indicated a good precision and accuracy for the proposed UPLC-MS/MS method (Table 4).
Table 4 Determination of vildagliptin, sitagliptin and metformin in laboratory prepared mixtures and in Galvus Met and Janumet® tablets (application of standard addition technique) by the proposed LC-MS\MS methoda
| Item |
Laboratory prepared mixtures |
| Concentration (ng mL−1) |
% recovery* |
| VLG |
MET |
STG |
VLG |
MET |
STG |
| SD = standard deviation, % RSD = percent relative standard deviation, *average of three different determinations, **ratio present in Galvus Met® tablets, ***ratio present in Janumet® tablet. |
| |
300.00 |
300.00 |
30.00 |
98.98 |
100.35 |
100.25 |
| 25.00 |
50.00 |
25.00 |
100.33 |
99.54 |
98.95 |
| 75.00 |
375.00 |
75.00 |
99.35 |
100.38 |
100.84 |
| **20.00 |
200.00 |
— |
101.00 |
98.89 |
— |
| 200.00 |
100.00 |
100.00 |
98.87 |
100.99 |
99.12 |
| — |
400.00 |
***20.00 |
— |
101.15 |
99.50 |
| Mean* ± SD |
|
99.71 ± 0.92 |
100.22 ± 0.86 |
99.73 ± 0.80 |
| % RSD |
0.93 |
0.86 |
0.80 |
| Variance |
0.85 |
0.74 |
0.63 |
| Item |
Galvus Met® tablets |
Janumet® tablets |
| Taken concentration (ng mL−1) |
Added concentration (ng mL−1) |
% recovery* |
Taken concentration (ng mL−1) |
Added concentration (ng mL−1) |
% recovery* |
| VLG |
MET |
VLG |
MET |
VLG |
MET |
STG |
MET |
STG |
MET |
STG |
MET |
| |
5.00 |
50.00 |
15.00 |
100.00 |
99.87 |
99.87 |
5.00 |
100.00 |
5.00 |
100.00 |
98.89 |
100.55 |
| 45.00 |
250.00 |
98.55 |
99.48 |
10.00 |
200.00 |
99.54 |
100.17 |
| 95.00 |
150.00 |
101.25 |
100.75 |
20.00 |
150.00 |
98.24 |
99.16 |
| 195.00 |
300.00 |
99.69 |
100.98 |
35.00 |
300.00 |
100.46 |
99.59 |
| 295.00 |
50.00 |
100.47 |
99.24 |
75.00 |
300.00 |
98.34 |
99.85 |
| Mean* ± SD |
99.39 ± 0.88 |
99.12 ± 0.78 |
|
100.16 ± 1.07 |
100.06 ± 0.77 |
98.89 ± 0.65 |
99.10 ± 0.81 |
|
|
99.09 ± 0.92 |
99.86 ± 0.53 |
| % RSD |
0.88 |
0.79 |
|
1.07 |
0.77 |
0.66 |
0.82 |
0.93 |
0.53 |
| Variance |
0.77 |
0.61 |
|
1.14 |
0.59 |
0.42 |
0.66 |
0.85 |
0.28 |
Beside the analysis of dosage forms, preliminary study was carried out to test the ability of the proposed method to analyze plasma samples. Thus, plasma samples were spiked with different concentrations of the standards of the analytes and deproteinization was carried out by the addition of acetonitrile. The accuracy and precision were evaluated by analyzing replicate spiked samples at different concentrations. The obtained primary results has proved the proposed method to be suitable for the determination of STG, VLG and MET in spiked plasma samples with accuracy of 96.87, 95.98% and 96.02% and precision of 0.93, 1.36 and 1.10, respectively. As a future work; a complete validation for the method will be conducted according to Guidance for Industry Bioanalytical Method Validation, recommended by FDA.30 In addition real plasma samples from human volunteers will be analyzed to judge the potential of our method for bioavailability studies.
3.4. Degradation behavior
During stability studies, 20–80% degradation of the substance to be examined is to be achieved to qualify the assay method as being able to indicate stability.31 The studied drugs; STG, VLG and MET were subjected to acidic, basic, thermal and oxidative degradation. Several degradation products were detected in the full scan spectra of [M + H]+ of the degradation solutions of the three drugs (Fig. 4). From our study it was obvious that STG was susceptible to both acidic and alkaline degradation but resistant to thermal and oxidative degradation. The degradation pathways of STG and structure elucidation of the main degradates are represented in Scheme 1. On the other hand, VLG was extensively degraded under basic conditions followed by oxidative conditions and to a lesser extent it was affected by both acidic and thermal conditions. It is worth noting that there was only one main degradation pathway for VLG under all applied stress conditions which is the cleavage of the secondary amine with the production of primary amine degradates (Scheme 2). Finally, MET was very rapidly degraded under the utilized basic conditions, followed by the thermal conditions. Structure elucidation of the produced MET degradates are shown Scheme 2.
 |
| | Fig. 4 Full scan spectra of [M + H]+ of the degradation products of: (a) MET, (b) VLG. (c) STG acid degradation products and (d) STG alkaline degradation products. | |
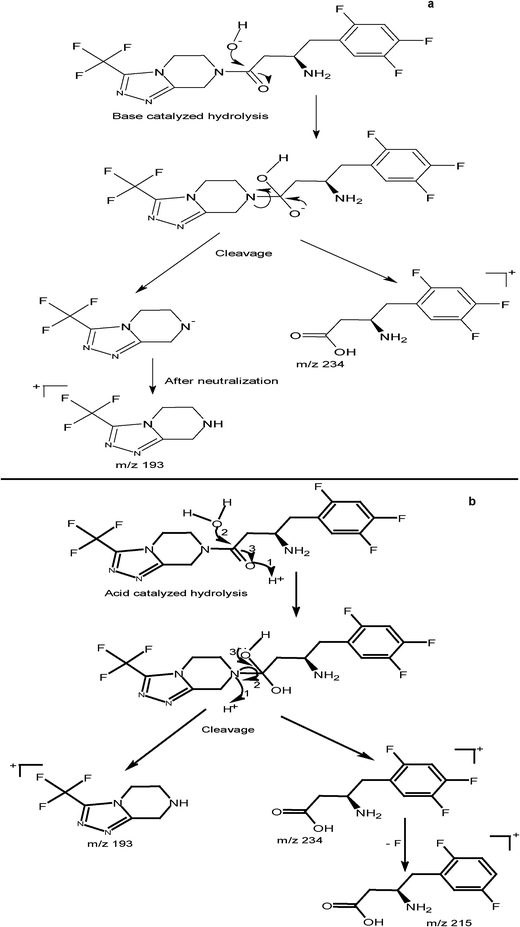 |
| | Scheme 1 The proposed structures and pathways of the main degradation products of STG produced under (a) alkaline degradation and (b) acid degradation. | |
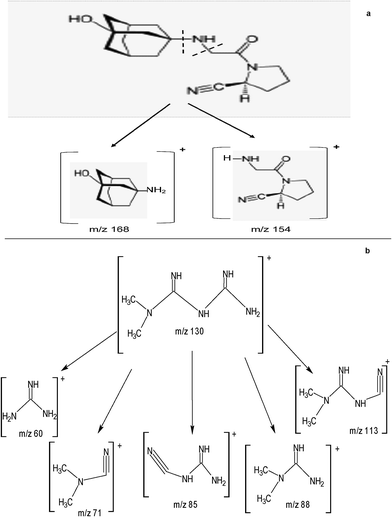 |
| | Scheme 2 The proposed structures of the main degradation products of: (a) VLG and (b) MET. | |
At this stage we would like to draw the attention to the similarity between some of the degradation products produced by our method when subjecting VLG and MET to the different stress conditions and the product ions produced in the Q3 when performing MS/MS of VLG and MET by subjecting the drugs to the collision energy. From these were m/z 154 for VLG and m/z 71 for MET.
Some authors have performed different stress studies on STG,11,12,32 VLG33 and MET.12 In order to find out the effect of the differences in stress conditions applied on the drugs and the produced degradates we have performed a comparison between our stress studies and other reported ones (specifically reported methods which have elucidated the structures of the produced degradates) as demonstrated in Table 5. The percentage degradation of the three drugs as well as the protonated molecular masses of the main degradation products are illustrated in Table 5. As expected the percentage of the produced degradation products was greatly affected by the conditions, the more severe the conditions, the more rapid was the degradation and the higher the percentage of the degradation. However, the differences in the applied conditions from one method to another were a bit confusing. Regarding STG and VLG although the applied conditions were different between our proposed method and the reported ones11,12,32,33 however, in most cases the produced degradates were similar, which proves the validity of the degradation pathways introduced by us in Schemes 1 and 2. On the contrary, the degradation products of MET produced by our proposed method and by Peraman et al.,12 were completely different.
Table 5 Comparison between the stress conditions applied by the proposed method and some reported methods11,12,32,33 showing the percentage degradation of the three drugs and the protonated molecular masses of the main degradation products
| Drug |
Method of degradation |
| |
Method |
Conditions and percentage degradation |
[M + H]+ of the degradation products |
| STG |
Acid hydrolysis |
Proposed |
Heating in boiling water bath with 1.0 N HCl for 1 h |
m/z 215 (the most abundant), 113, 155 and 234 |
| Degradation reached 40% after 30 min and 68% after 1 h |
(Structure elucidation in Scheme 1) |
| El-Bagary et al.32 |
Heating in boiling water bath with 6.0 N HCl for 8 h |
m/z 234 |
| Degradation reached 100% |
3-Amino-4-(2,4,5 trifluorophenyl)butanoic acid |
| Peraman et al.12 |
Treating with 1 M HCl at room temperature for 48 h |
m/z 215 |
| Degradation reached 16.58% |
(R)-3-Amino-4-(2,5-difluorophenyl)butanoic acid |
| Base hydrolysis |
Proposed |
Heating in boiling water bath with 1.0 N NaOH for 2 h |
m/z 193 (the most abundant), 113, 155, 215, 227, and 249 |
| Degradation reached 68% after 30 min, and 93% after 2 h |
(Structure elucidation in Scheme 1) |
| El-Bagary et al.11 |
Refluxing in a boiling water bath with 5 N NaOH for 6 h |
m/z 193 |
| Degradation reached 100% |
|
| Peraman et al.12 |
Treating with 0.1 N NaOH at room temperature for 48 h |
m/z 215 |
| Degradation reached 64.27% |
(R)-3-Amino-4-(2,5-difluorophenyl)butanoic acid |
| Thermal hydrolysis |
Proposed |
Heating with deionized water in boiling water bath for 2 h |
Negligible degradation |
| Peraman et al.12 |
Dry heating in an oven at 105 °C for 6 h |
m/z 215 + other unidentified products |
| Degradation reached 60.5% |
(R)-3-Amino-4-(2,5-difluorophenyl)butanoic acid |
| Oxidative hydrolysis |
Proposed |
Treating with 0.6% hydrogen peroxide for 2 h |
Negligible degradation |
| Peraman et al.12 |
Treating with 0.3% H2O2 for 5 days |
Negligible degradation |
| VLG |
Acid hydrolysis |
Proposed |
Heating in boiling water bath with 1.0 N HCl for 2 h |
m/z 154 and 168 (structure elucidation in Scheme 2) |
| Barden et al.33 |
Treating with 1 M HCl at room temperature for 24 h |
Negligible degradation |
| Base hydrolysis |
Proposed |
Heating in boiling water bath with 1.0 N NaOH for 1 h |
m/z 154 and 168 (structure elucidation in Scheme 2) |
| Degradation reached 99.61% after 1 h |
|
| Barden et al.33 |
Treating with 0.1 M NaOH, at room temperature, for 2 h |
m/z 154 |
| Thermal hydrolysis |
Proposed |
Heating with deionized water in boiling water bath for 2 h |
m/z 154 (structure elucidation in Scheme 2) |
| Barden et al.33 |
Heating at 60 °C for 240 h |
m/z 154 |
| Oxidative hydrolysis |
Proposed |
Treating with 0.6% hydrogen peroxide for 1 h |
m/z 154 (structure elucidation in Scheme 2) |
| Degradation reached 92.78% after 1 h |
|
| Barden et al.33 |
Treating with 0.3% hydrogen peroxide for 2 h |
m/z 154 |
| MET |
Acid hydrolysis |
Proposed |
Heating in boiling water bath with 1.0 N HCl for 2 h |
m/z 60 and 71, 85, 88 and 113 (structure elucidation in Scheme 2) |
| Peraman et al.12 |
Treating with 1 M HCl at room temperature for 48 h |
Negligible degradation |
| Base hydrolysis |
Proposed |
Heating in boiling water bath with 1.0 N NaOH for 1 h |
m/z 60 and 71, 85, 88 and 113 with m/z 71 and 60 as the most abundant ones (structure elucidation in Scheme 2) |
| Degradation reached 99.78% after 10 min |
|
| Peraman et al.12 |
Treating with 0.1 N NaOH at room temperature for 48 h |
m/z 116 |
| Degradation reached 9.88% |
1-(Diaminomethylidene)-3-methylguanidine |
| m/z 127 |
| 1,3,5-Triazine-2,4,6-triamine |
| Thermal hydrolysis |
Proposed |
Heating with deionized water in boiling water bath for 2 h |
m/z 60 and 71, 85, 88 and 113 with m/z 71 and 60 as the most abundant ones. (Structure elucidation in Scheme 2) |
| Degradation reached 54.88% after 1 h |
|
| Peraman et al.12 |
Dry heating in an oven at 105 °C for 6 h |
m/z 127 |
| Degradation reached 20.57% |
1,3,5-Triazine-2,4,6-triamine |
| Oxidative hydrolysis |
Proposed |
Treating with 0.6% hydrogen peroxide for 1 h |
m/z 60 and 71, 85, 88 and 113 |
| (Structure elucidation in Scheme 2) |
| Peraman et al.12 |
Treating with 0.3% H2O2 for 5 days |
Negligible degradation |
3.5. Degradation kinetics
The kinetics of alkaline and acidic degradations of STG, alkaline and oxidative degradations of VLG and alkaline and thermal degradations of MET was investigated through withdrawing different samples of the degradation solutions at different time intervals (5, 10, 15, 30, 60, 120 min). A regular decrease in the drugs concentrations was detected with increasing time. When plotting the log of remained drugs concentration versus time, a linear relationship was observed with good correlation coefficients (Fig. 5).
 |
| | Fig. 5 First order plots for the degradation of: (a) VLG under: (a1) alkaline degradation and (a2) oxidative degradation, (b) MET under: (b1) alkaline degradation and (b2) thermal degradation and (c) STG under: (c1) acidic degradation and (c2) alkaline degradation. | |
Pseudo-first-order is the term used when two reactants are involved in the reaction but one of them is in such a large excess that any change in its concentration is negligible compared with the change in the concentration of the other reactant (drug). Rate constant (K), time left for 50% potency (t1/2) and time left for 90% potency (t90) under alkaline and acidic stress conditions were calculated using eqn (1)–(3), respectively:34
| |
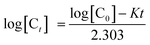 | (1) |
| |
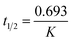 | (2) |
| |
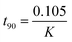 | (3) |
where
K is the rate constant, [C
0] is the concentration of STG or VLG or MET at time
t = 0 and [C
t] is its concentration at time
t. The
K values per minute were found to be 4.7 × 10
−1 and 7.6 × 10
−1, while
t1/2 values were 1.47 and 0.91 min and
t90 values were 0.22 and 0.14 min for acidic and alkaline degradations of STG, respectively. Additionally, the
K values per minute were found to be 2.0 × 10
−1 and 4.0 × 10
−2, while
t1/2 values were 3.25 and 15.83 min and
t90 values were 0.49 and 2.39 min for alkaline and oxidative degradations of VLG, respectively. However, the
K values per minute were found to be 6.0 × 10
−1 and 3.0 × 10
−2, while
t1/2 values were 1.13 and 19.25 min and
t90 values were 0.17 and 2.91 min for alkaline and thermal degradations of MET, respectively. Extensive degradation was observed under alkaline stress conditions for the three drugs, where the
K values were found to be the highest. On contrary, both
t1/2 and
t90 at alkaline conditions were found to be lower than those of other conditions.
4. Conclusion
A novel, simple and specific stability-indicating UPLC-MS/MS method was developed for the simultaneous determination of sitagliptin (STG), vildagliptin (VLG) and metformin (MET) in pharmaceutical dosage forms in the presence of STG related impurities. The utilization of UPLC has enabled the use of a short column with small particle size which has resulted in well-defined chromatographic peaks within a very short run time. In addition the low flow rate applied in this method has offered less solvent consumption which is considered to be cost effective and eco-friendly. The proposed method has proved to be of high sensitivity with LOQ of 5.00 ng mL−1, 5.00 ng mL−1 and 10.00 ng mL−1 for STG, VLG and MET, respectively. The method was validated in accordance with ICH guidelines. The results gained from the validation study have confirmed that the new method is selective, linear, precise and accurate. Additionally, the proposed method is specific and unaffected by the presence of degradation products from stress degradation and STG impurities, confirming the stability-indicating nature of the method. Moreover, for the first time degradation kinetics of the drugs was also studied by our method proving that degradation was a pseudo-first-order reaction, and it was concluded STG, VLG and MET were extensively degraded under basic conditions. Based on all previous advantages and results the developed method can be conveniently used by quality control laboratories.
References
- C. B. Sekaran and A. P. Rani, Int. J. Pharm. Pharm. Sci., 2010, 2, 138–142 Search PubMed.
- R. Nirogi, V. Kandikere, K. Mudigonda, P. Komarneni, R. Aleti and R. Boggavarapu, Biomed. Chromatogr., 2008, 22, 214–222 CrossRef CAS PubMed.
- W. Zeng, D. Musson, A. L. Fisher, L. Chen, M. S. Schwartz, E. Woolf and A. Q. Wang, J. Pharm. Biomed. Anal., 2008, 46, 534–542 CrossRef CAS PubMed.
- M. M. Jost, J. Lamerz, H. Tammen, C. Menzel, I. De Meester, A. M. Lambeir, K. Augustyns, S. Scharpé, H. D. Zucht, H. Rose, M. Jürgens, P. Schulz-Knappe and P. Budde, Biochem. Pharmacol., 2009, 77, 228–237 CrossRef CAS PubMed.
- P. Sengupta, U. Bhaumik, A. Ghosh, A. K. Sarkar, B. Chatterjee, A. Bose and T. K. Pal, Chromatographia, 2009, 69, 1243–1250 CAS.
- V. Souza-Mello, B. M. Gregório, F. S. Cardoso-de-Lemos, L. De Carvalho, M. B. Aguila and C. A. Mandarim-de-Lacerda, Clin. Sci., 2010, 119, 239–250 CrossRef CAS PubMed.
- The British Pharmacopoeia, The Stationery Office, Electronic version, London, 2009, pp. 3813–3816 Search PubMed.
- The United States Pharmacopoeia 30, the National Formulary 25, US Pharmacopeial Convention, Electronic version, Rockville, MD, 2007 Search PubMed.
- R. I. El-Bagary, E. F. Elkady and B. M. Ayoub, Int. J. Biomed. Sci., 2011, 7, 62–69 CAS.
- G. Khan, D. Sahu, Y. P. Agrawal, N. Sabarwal, A. Jain and A. K. Gupta, sian J. Biochem. Pharm. Res., 2011, 1, 352–358 CAS.
- R. I. El-Bagary, E. F. Elkady and B. M. Ayoub, Talanta, 2011, 85, 673–680 CrossRef CAS PubMed.
- R. Peraman, C. S. Gowra, Y. P. Reddy and K. K. Peruru, Chromatographia, 2013, 76, 1153–1162 CAS.
- S. A. Kumar, M. Debnath and J. V. L. N. S. Rao, Pharm. Sin., 2013, 4, 47–61 CAS.
- P. V. Diwan, N. K. Surendra, H. Sanayaima, N. Anusha and V. V. L. N. Prasad, International Journal of Pharmacy and Industrial Research, 2012, 2, 283–290 Search PubMed.
- C. S. N. Malleswararao, V. M. Suryanarayana and K. Mukkanti, Sci. Pharm., 2012, 80, 139–152 CrossRef CAS PubMed.
- R. I. El-Bagary, E. F. Elkady and B. M. Ayoub, Eur. J. Chem., 2013, 4, 444–449 CrossRef CAS PubMed.
- J. Martín, W. Buchberger, J. L. Santos, E. Alonso and I. Aparicio, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 895–896, 94–101 CrossRef PubMed.
- S. L. Bonde, R. P. Bhadane, A. Gaikwad, D. Katale, S. Gavali and A. S. Narendiran, Int. J. Pharm. Pharm. Sci., 2013, 5, 463–470 CAS.
- M. Salim, N. El-Enany, F. Belal, M. Walash and G. Patonay, Anal. Chem. Insights, 2012, 7, 31–46 CrossRef CAS PubMed.
- M. F. Abdel-Ghany, O. Abdel-Aziz, M. F. Ayad and M. M. Tadros, Spectrochim. Acta, Part A, 2014, 125, 175–182 CrossRef CAS PubMed.
- P. S. R. C. H. N. P. Varma, A. L. Rao and S. C. Dinda, Int. Res. J. Pharm., 2013, 4, 122–128 Search PubMed.
- B. Santhosha, A. Ravindranath and C. Sundari, Int. Res. J. Pharm. Appl. Sci., 2012, 2, 22–28 CrossRef CAS.
- U. Gundala, C. S. Bhuvanagiri and D. Nayakanti, indo american journal of pharmaceutical research, 2013, 3, 1554–1563 Search PubMed.
- M. A. Mohammad, E. F. Elkady and M. A. Fouad, Eur. J. Chem., 2012, 3, 152–155 CrossRef CAS.
- R. Pontarolo, A. C. Gimenez, T. M. de Francisco, R. P. Ribeiro, F. L. Pontes and J. C. Gasparetto, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 965, 133–141 CrossRef CAS PubMed.
- C. P. Uber, F. L. D. Pontes, J. C. Gasparetto, T. M. G. De Francisco, M. S. Piantavini, M. A. Cardoso and R. Pontarolo, Int. J. Pharm. Pharm. Sci., 2014, 6, 203–207 CAS.
- ICH Harmonized Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2(R1), Current step 4 version, Parent guidelines on Methodology, 1996, Incorporated in November 2005.
- ICH Harmonised Tripartite Guidelines, Q1A(R2) Stability Testing of New Drug Substances and Products, 2003.
- J. N. Miller and J. C. Miller, Statistics and Chemometrics for Analytical Chemistry, Pearson Education Limited, England, Harlow, 5th edn, 2005 Search PubMed.
- FDA, Guidance for industry: bioanalytical method validation, US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CV), 2001.
- R. Sehrawat, M. Maithani and R. Singh, Chromatographia, 2010, 72, 1–6 CAS.
- R. I. El-Bagary, E. F. Elkady and B. M. Ayoub, Eur. J. Chem., 2013, 4, 360–365 CrossRef CAS.
- A. T. Barden, B. Salamon, E. E. S. Schapoval and M. Steppe, J. Chromatogr. Sci., 2012, 50, 426–432 CAS.
- K. A. Connors, G. R. Amidon and V. J. Stella, A Handbook for Pharmacists, Wiley, New York, 2nd edn, 1986 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.